顶空进样法检测氟胞嘧啶中的残留溶剂
顶空进样-气相色谱法测定植物提取物中17种有机溶剂残留

顶空进样-气相色谱法测定植物提取物中17种有机溶剂残留作者:胡凤杨来源:《现代食品》 2017年第5期植物提取物是指将植物中某一种或多种有效成分经过物理或化学方法提取、分离和浓缩所形成的产品,常用作药品、保健食品、化妆品等的原料或添加成分[1]。
在萃取和富集的过程中,会用到多种有机溶剂,如甲醇、乙醇、乙酸乙酯等[2],有时还会用到大孔树脂吸附技术,而大孔树脂在制备的过程中会使用苯、甲苯、二乙烯苯等有机溶剂[3],如果不能完全去除这些有机溶剂,就会造成植物提取物中有多种有机溶剂残留,进而导致由植物提取物制成的产品也有有机溶剂残留。
由于大部分有机溶剂对人体有害,因此建立一套植物提取物中同时检测多种有机溶剂残留的方法十分有必要。
本文以葡萄籽提取物为样品,建立了基于顶空进样法测定植物提取物中17 种有机溶剂残留的气相色谱分析方法,对前处理方法、顶空进样参数和气相色谱仪分析条件分别进行了优化,并对该方法进行了方法学的考察。
1 材料与方法1.1 仪器与试剂1.1.1 仪器丹尼HSS 86.50 顶空进样器、安捷伦7820A 气相色谱仪、安捷伦HP-INNOWax 毛细管柱(60 m×0.25 mm×0.5 μm)、安捷伦20 mL 顶空进样瓶和Milipore 纯水机。
1.1.2 试剂实验用水为去离子水,N,N- 二甲基甲酰胺(DMF)及其余试剂均为分析纯,广州化学试剂厂;葡萄籽提取物。
1.2 方法1.2.1 标准曲线的配制将各有机溶剂配制成约10 mg/mL 的储备液,用DMF 定容。
根据各溶剂在气相色谱仪上的响应值大小分别吸取不同体积的储备液配制成混合标准中间工作液,用50%DMF 水溶液定容。
最后将混合标准中间工作液稀释配制成一系列标准溶液,用50%DMF 水溶液定容至10 mL,量取5 mL 置顶空进样瓶,密封。
1.2.2 样品的处理称取约0.2 g 样品,用50%DMF 水溶液定容至10 mL,超声5 min,取5 mL 置顶空进样瓶,密封。
顶空进样-气相色谱法测定植物提取物中17种有机溶剂残留

顶空进样-气相色谱法测定植物提取物中17种有机溶剂残留胡凤杨
【期刊名称】《粮食流通技术》
【年(卷),期】2017(005)010
【摘要】建立测定植物提取物中17种有机溶剂残留的检测方法.将植物提取物用50%DMF水溶液溶解,采用顶空进样-气相色谱法检测17种有机溶剂残留.结果显示:各有机溶剂相关系数均大于0.99,精密度在2.0%~6.3%,除正己烷、四氢呋喃、乙酸乙酯三个有机溶剂的回收率大于70%外,其余有机溶剂的低、中、高加标水平回收率均大于80%,各有机溶剂的检出限均远低于中国药典(2015年版)规定的限量值.本方法操作简单,稳定、准确、灵敏,适用于植物提取物中17种有机溶剂残留检测.
【总页数】5页(P124-128)
【作者】胡凤杨
【作者单位】中国广州分析测试中心,广东广州 510070
【正文语种】中文
【中图分类】O657.7+1
【相关文献】
1.顶空进样-气相色谱法测定盐酸沙格雷酯原料药中有机溶剂残留 [J], 郭捷;庞小刚
2.顶空进样气相色谱法同时测定DP中7种有机溶剂残留量 [J], 梅黎;崔丽霞
3.顶空进样-气相色谱法测定植物提取物中17种有机溶剂残留 [J], 胡凤杨;
4.顶空进样气相色谱法测定葛根提取物中有机溶剂残留研究 [J], 刘媛;陈丽
5.顶空进样气相色谱法测定化学合成常用四种有机溶剂残留 [J], 田辉;张明
因版权原因,仅展示原文概要,查看原文内容请购买。
顶空气相色谱法测定化妆品中15种挥发性有机溶剂残留

c e ty f o m purte ih h g s nstv t nd r pr ducbiiy. The r l tve s a i n l r m i ii s w t i h e ii iy a e o i lt e a i t nda d d v a i ns r e i to
( ne  ̄o s a eC nto n Ce tr rDie s o r l d n例 t n o a g n o i c , f a i fGu n  ̄ gPr vn e Gua g h u 5 ∞D ≮C i a o n z o 1 D h n )
Ab t a t s r c :A e d p c a h o ao r p y ( ・ h a s a eg sc r m t ga h HS GC)m e h d w a e eo e o h a i e t o sd v tp d f rt e r p d d —
t r i ton o ulim i ur 5 r sdua l tl ga c c e m na i fm t— xt e of1 e i l vo a ie or ni om pou nds i os e i s The e n c m tc . xpe - r i e a o m nt lc ndii ns ofH S— to GC uc ad p e t m pe a u e,he ds s h ashe s ac e rtr a pac i e a he a l tc l e tm nd t na y i a c dii s o on ton f GC e e o i ied. Cos e i a pl s w e e e r c e nd c ea ed up b a w r ptm z m tc s m e r xta t d a l n — y he d—
顶空气相色谱法测定中药提取物的有机溶剂残留量

顶空气相色谱法测定中药提取物的有机溶剂残留量
石云峰;金樟照
【期刊名称】《中国药业》
【年(卷),期】2007(16)19
【摘要】目的建立中药提取物XDD-2的有机溶剂正丁醇、乙醇和石油醚残留量检查法.方法用顶空气相色谱法测定XDD-2的有机溶剂残留量,采用HP-5固定相的开口毛细管柱,以高纯氮为载气,FID为检测器.结果标准液质量浓度在一定范围内与各组分峰面积线性关系艮好,R2在0.990 9~0.999 9之间,精密度、重现性RSD 均小于5%,平均回收率为95%~105%.结论顶空气相色谱法简便,快速,准确,可用于中药制剂的正丁醇、乙醇和石油醚残留量测定.
【总页数】2页(P12-13)
【作者】石云峰;金樟照
【作者单位】浙江省药品检验所,浙江,杭州,310004;浙江省药品检验所,浙江,杭州,310004
【正文语种】中文
【中图分类】R927.11
【相关文献】
1.顶空气相色谱法测定荷叶提取物中11种有机溶剂的残留量 [J], 黄媛
2.顶空气相色谱法检测苦荞麦总黄酮提取物的有机溶剂残留量 [J], 邹亮;杨敬东;赵钢;罗亚玲
3.顶空气相色谱法同时测定黄蜀葵花总黄酮提取物中9种有机溶剂的残留量 [J], 张清华; 张治云; 杨海霞
4.顶空气相色谱法测定奥贝胆酸原料药中有机溶剂的残留量 [J], 付丙月;吴伟山;张宁;王金虎
5.顶空气相色谱法测定盐酸奥布卡因中有机溶剂残留量 [J], 魏乐坤;张肖肖;刘宜辉因版权原因,仅展示原文概要,查看原文内容请购买。
顶空气相色谱法测定富马酸卢帕他定中9种有机溶剂残留量

顶空气相色谱法测定富马酸卢帕他定中9种有机溶剂残留量张雷;靳守东;王晓玲;卢燕【摘要】目的:测定富马酸卢帕他定中9种有机溶剂残留量.方法:采用气相色谱法,PE-9000气相色谱仪、PE TurboMatrix 16顶空进样器、氢火焰检测器、DB-624(30 m×0.32 mm×1.8 μm)石英毛细管柱.结果:连续进样6针,精密度良好,在考察的浓度范围内均呈良好的线性关系,回收率均符合要求.结论:方法稳定,操作简便、准确、可靠,适用于富马酸卢帕他定中9种有机溶剂残留量的检测.【期刊名称】《中国医院用药评价与分析》【年(卷),期】2011(011)001【总页数】2页(P48-49)【关键词】富马酸卢帕他定;气相色谱法;有机溶剂残留量【作者】张雷;靳守东;王晓玲;卢燕【作者单位】首都医科大学附属北京儿童医院,北京,100045;中国人民解放军总后勤部卫生部药品仪器检验所,北京,100071;首都医科大学附属北京儿童医院,北京,100045;首都医科大学附属北京儿童医院,北京,100045【正文语种】中文【中图分类】R976富马酸卢帕他定(fumarate rupatadine)是西班牙Uriach制药公司研制的具抗组胺和抗血小板活化因子(PAF)双重作用的抗过敏药物,适应证为季节性和常年性过敏性鼻炎。
PAF可引起支气管的收缩和血管通透性的增强,从而导致流涕和鼻充血。
目前,临床上使用的抗过敏药均只有抗组胺活性,而没有PAF拮抗作用,卢帕他定是既具有抗阻胺作用又拮抗PAF活性的抗过敏药[1,2]。
由于其生产工艺使用到甲醇、乙醇、乙腈、二氯甲烷、正己烷、乙酸乙酯、四氢呋喃、氯仿、甲苯9种溶剂,故须对此9种溶剂的残留量加以控制。
本文采用顶空气相色谱法同时对9种残留溶剂进行测定[3],操作简便,重现性好,结果准确可靠。
1 仪器与试药PE-9000气相色谱仪、PE TurboMatrix 16顶空进样器、FID检测器;富马酸卢帕他定为国内仿制产品,批号为090316、090318、090319,所用试剂均为分析纯。
顶空-气相色谱法测定食用植物油中溶剂残留的方法研究
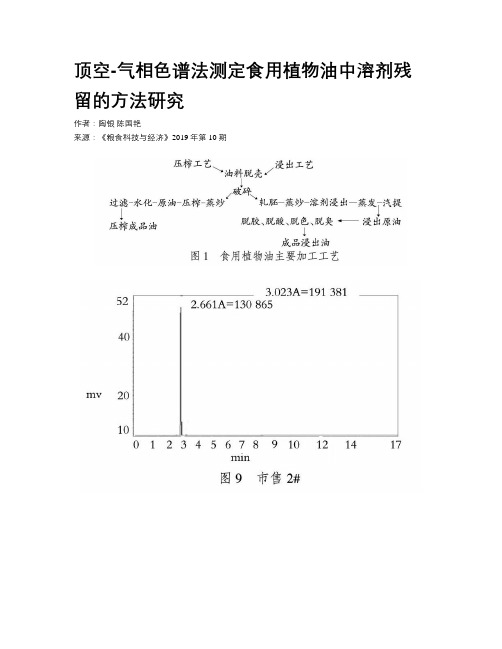
顶空-气相色谱法测定食用植物油中溶剂残留的方法研究作者:陶银陈国艳来源:《粮食科技与经济》2019年第10期[摘要]建立一种快速、准确、操作简单的通过顶空-气相色谱法测定食用植物油中溶剂残留的分析方法,在国标基础上,对顶空进样器、色谱柱测定条件进行分析验证,采用内标法进行定量,得出该方法灵敏度高,并且能简便、准确、快速地测定出食用植物油中的溶剂残留。
[关键词]顶空-气相色谱法;食用植物油;溶剂残留中图分类号:O657.71;TS227 文献标识码:A DOI:10.16465/431252ts.201910食用植物油是食用油的一种,即植物性油料作物的含油部分经过压榨、精炼等工艺而成的食用油品,油料作物经过初榨产出原油和粕类(副产品),原油精炼后,即为食用植物油。
植物油是人体必需脂肪酸的重要来源,能提供人体必需的热量,且有利于维生素A、D等脂溶性维生素的吸收,对于保证人体健康和营养平衡很有很大帮助。
随着居民生活水平的不断提高,健康观念深入人心,作为人们日常消费的必需品之一,国内食用植物油的消费观念和结构也发生了变化,人们对食用油的品质要求也日渐苛刻,在满足需求的同时,对食用油的营养和品质以及安全性也有了一定的要求。
1 食用油加工工艺分析食用植物油的加工工艺主要有两种:直接压榨法和溶剂浸出法[1-2](见图1)。
由图1浸出油工艺分析可知,需要加入溶剂浸提,再加上脱胶、脱酸、脱色、脱臭才能得到成品浸出油。
虽然工艺中会去除很大一部分溶剂,可仍然会有溶剂残留;而在我国,为了提高出油率,对于压榨后的油渣油饼,仍然还会用溶剂再次进行浸提,用溶剂浸提,成品油里就会有少量残留的溶剂[3-4]。
浸出法常用的溶剂为6号溶剂,最主要的成分为正己烷,但正己烷挥发性强、脂溶性高,易在体内蓄积并产生神经毒性[5-9],因此我国常见的植物油均把溶剂残留作为强制性指标加以检验;溶剂残留是食用植物油卫生标准的必检项目,其标准规定值为原油100mg/kg、植物油为50mg/kg[10-14]。
-顶空气相色谱-质谱联用法测定食用植物油中的溶剂残留

-顶空气相色谱-质谱联用法测定食用植物油中的溶剂残留王少伟【期刊名称】《食品安全导刊》【年(卷),期】2015(000)021【总页数】3页(P98-100)【作者】王少伟【作者单位】江苏省食品药品监督检验研究院【正文语种】中文食用植物油的加工工艺分为压榨法与浸出法。
浸出法因具有粕饼中含残油少、出油率高、加工成本低、经济效益高等优点,被国内外植物油生产厂家广泛采用。
然而,植物油中萃取剂—六号溶剂的残留也变得不可避免。
六号溶剂是一种以六碳烷烃为主要成分的烷烃、环烷烃、芳烃等各种低级烷烃混合物,其中所含少量芳烃及硫化物杂质有较大毒性,长期接触会麻醉呼吸中枢、损伤皮肤屏障功能、损害周围神经和造血功能。
研究食用植物油中溶剂残留检测方法,加强对食用植物油中溶剂残留量的监控,有利于提高我国食用油安全品质,保障人民生命财产安全。
国内对于食用植物油中溶剂残留顶空-气相色谱检测方法研究较广泛。
汪海峰、鞠兴荣等对食用植物油中溶剂残留的高温顶空-气相色谱测定方法进行了优化;喻利娟、史玉坤研究了溶剂基质对六号溶剂测定的影响。
目前,国内溶剂残留检测器大多选用FID检测器。
质谱MS检测器作为一种通用型检测器,在食用植物油溶剂残留检验中的应用报道仍然较少,食用植物油溶剂残留气相色谱-质谱联用检测的定量方法更是几乎没有。
通过气相色谱-质谱联用获得的化合物质谱图进行谱库检索,可以有效弥补了气相色谱定性的缺陷。
MS多种扫描方式可以有效排除基质和杂质峰的干扰,通过全扫描模式(SCAN)确定组分特征离子,再对化合物特征离子(SIM)扫描,以面积归一法建立定量方法,可以有效提高检测灵敏度,并提升检验结果的准确性。
本文应用顶空气相-质谱联用技术分析食用植物油中溶剂残留,并建立基于顶空气相-质谱联用条件下的定量分析技术,以期为食用植物油的品质评价提供更准确的依据。
实验材料六号溶剂标准溶液来源:国家粮食局科学院;食用植物油(样品)来源:市场抽样;新鲜机榨油来源:南京市江宁植物油厂;仪器与试剂实验试剂N,N-二甲基乙酰胺(DMF)(化学纯级)中国医药集团上海化学试剂公司。
0861残留溶剂测定法

0861残留溶剂测定法药品中的残留溶剂系指在原料药或辅料的生产中,以及在制剂制备过程中使用的,但在工艺过程中未能完全去除的有机溶剂。
药品中常见的残留溶剂及限度见附表1,除另有规定外,第一、第二、第三类溶剂的残留限度应符合附表1中的规定;对其他溶剂,应根据生产工艺的特点,制定相应的限度,使其符合产品规范、药品生产质量管理规范(GMP)或其他基本的质量要求。
本法一般采用色谱法,如照气相色谱法(通则0521)测定。
色谱柱1. 毛细管柱除另有规定外,极性相近的同类色谱柱之间可以互换使用。
(1)非极性色谱柱固定液为100%的二甲基聚硅氧烷的毛细管柱。
(2)极性色谱柱固定液为聚乙二醇(PEG-20M)的毛细管柱。
(3)中极性色谱柱固定液为(35%)二苯基-(65%)甲基聚硅氧烷、(50%)二苯基-(50%)二甲基聚硅氧烷、(35%)二苯基-(65%)二甲基聚硅氧烷、(14%)氰丙基苯基-(86%)二甲基聚硅氧烷、(6%)氰丙基苯基-(94%)二甲基聚硅氧烷的毛细管柱等。
(4)弱极性色谱柱固定液为(5%)苯基-(95%)甲基聚硅氧烷、(5%)二苯基-(95%)二甲基硅氧烷共聚物的毛细管柱等。
2. 填充柱以直径为0.18~0.25mm的二乙烯苯-乙基乙烯苯型高分子多孔小球或其他适宜的填料作为固定相。
系统适用性试验(1)用待测物的色谱峰计算,毛细管色谱柱的理论板数一般不低于5000;填充柱的理论板数一般不低于1000。
(2)色谱图中,待测物色谱峰与其相邻色谱峰的分离度应大于1.5。
(3)以内标法测定时,对照品溶液连续进样5次,所得待测物与内标物峰面积之比的相对标准偏差(RSD)应不大于5%;若以外标法测定,所得待测物峰面积的RSD 应不大于10%。
供试品溶液的制备1. 顶空进样除另有规定外,精密称取供试品0.1~1g;通常以水为溶剂;对于非水溶性药物,可采用N,N-二甲基甲酰胺、二甲基亚砜或其他适宜溶剂;根据供试品和待测溶剂的溶解度,选择适宜的溶剂且应不干扰待测溶剂的测定。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
顶空进样法检测氟胞嘧啶中的残留溶剂
摘要:目的建立气相色谱顶空进样法检测氟胞嘧啶中的残留溶剂1,2二氯苯和N,N二甲基苯胺方法顶空进样瓶体积20ml,进样量6ul,样品定量环体积1ml,平衡温度90℃,定量环温度100℃,传输线温度110℃,平衡时间10min,检测周期25min。
结果1,2二氯苯和N,N二甲基苯胺的分离度为9.41;1,2-二氯苯峰面积的重复性为RSD=0.87%,N,N二甲基苯胺的峰面积重复性为RSD=1.66%;1,2-二氯苯峰面积的中间精密度RSD=0.28%,N,N-二甲基苯胺峰面积中间精密度RSD=1.53%;1,2二氯苯的线性关系为y=13.347x-0.2557r=0.9997,N,N二甲基苯胺的线性关系y=20.221x-0.0335 r=0.9998;1,2二氯苯的回收率为100.54%-101.15%,N,N二甲基苯胺的回收率为99.86%-100.60%;1,2-二氯苯的检测限为0.05909ug/ml,N,N-二甲基苯胺的检测限为0.03105ug/ml;1,2-二氯苯的定量限为0.2056ug/ml,N,N-二甲基苯胺的定量限为0.1548ug/ml;方法具备一定耐用性。
结论本方法分离度良好,准确度高,有很好的线性、检测限和定量限。
关键词顶空进样法氟胞嘧啶残留溶剂
氟胞嘧啶化学名为4-氨基-5-氟-2(1H)-嘧啶酮,是一种抗真菌药,用于治疗白念球菌和隐球菌等感染。
根据我公司氟胞嘧啶的工艺,产品氟胞嘧啶中的主要残留溶剂是1,2-二氯苯和N,N-二甲基苯胺,1,2-二氯苯的内控标准为50 ppm, N,N-二甲基苯胺的内控标准为5ppm,对残留溶剂1,2-二氯苯和N,N-二甲基苯胺的测试,现采用气相色谱顶空进样方法,有效地控制产品中的这两种残留溶剂,达到了满意的效果。
仪器与试药
安捷伦7890A气相色谱仪G1888顶空进样器安捷伦Chemstation 色谱软件DB-624(30m×0.53mm×3.0um)石英毛细管柱及氢火焰离子化检测器氟胞嘧啶工作标准品(FLC2010001S,本公司自制)1,2-二氯苯、N,N-二甲基苯胺和乙醇为色谱纯。
方法与结果
2.1【1】色谱条件
氮气作载气,柱流速4ml/min,进样器温度160℃,柱温150℃,检测器温度280℃,分流比:20:1,空气流速380ml/min,氢气流速30ml/min,尾吹+恒定柱流50ml/min。
顶空进样瓶体积20ml,进样量6ul,样品定量环体积1ml,平衡温度90℃,定量环温度100℃,传输线温度110℃,平衡时间10min,加压时间0.08min,定量环填充时间0.05min,定量环平衡时间0.05min,进梯时间
1.00min,检测周期25min。
2.2【1】样品和对照制备
2.2.1对照品溶液的制备(a)精确称取1,2-二氯苯0.0587克加入内有少量无水乙醇的100ml容量瓶中,用无水乙醇定容,即得标准贮备液(a)。
(b)精确称取N,N-二甲基苯胺0.0619克加入内有少量无水乙醇的100ml 容量瓶中,用无水乙醇定容,精确吸取该溶液1.00ml于10ml容量瓶中,用无水乙醇定容,即得标准贮备液(b)。
分别精确吸取标准贮备液(a)和标准贮备液(b)各1ml于100ml容量瓶中,定容,即得标准对照液。
此标准对照液相当于样品中1,2-二氯苯的浓度50ppm 左右,N.N-二甲基苯胺的浓度5ppm左右。
2.2.2 样品溶液的制备精确称取氟胞嘧啶样品1.0克,于10ml容量瓶中,用无水乙醇溶解后定容。
2.3【2】重复性
分别连续精确吸取(a)和(b)各1ml于10ml容量瓶中,用无水乙醇定容得标准对照溶液,吸取6ul分别进样。
六次测定的1,2-二氯苯的峰面积RSD=0.87%;N,N-二甲基苯胺的峰面积RSD=1.66%。
2.4【2】分离度
由重复性中任一张色谱图计算出1,2二氯苯和N,N二甲基苯胺的分离度为9.41。
2.5【2】中间精密度
将2.3中的标准对照溶液放置2天后,由不同的检验人员吸取6ul分别进样。
六次测定的1,2-二氯苯的峰面积RSD=0.28%;N,N-二甲基苯胺的峰面积RSD=1.53%。
2.6【2】线性
用标准贮备液(a)配制七种浓度的1,2-二氯苯测定其线性,得回归方程:y=13.347x-0.2557 ,相关系数(r):r=0.99997。
用标准贮备液(b)配制七种浓度的N,N-二甲基苯胺测定其线性,得回归方程:y=20.221x-0.0335相关系数(r):r=0.99963。
2.7【2】准确度(回收率)
准确称氟胞嘧啶工作标准品1.0g于10ml容量瓶中(三份),然后分别加入
2.2.1溶液(a)和溶液(b)0.7ml,1.0ml,1.3ml,配成三种浓度的样品。
每个浓度连续进样三针,进样量为6ul。
计算1,2-二氯苯各浓度的回收率分别是99.97%、100.52%、100.90%,RSD分别是0.04%、0.02%、0.24%;N,N二甲基苯胺的各浓度的回收率分别是99.13%、101.15%、99.44%,RSD分别是2.26%、0.54%、0.93%。
2.8【2】检测限定量限
在选定的色谱条件下,以信噪为在3:1为最低检测限进行测定,结果表明浓度为0.0587ug/ml的1,2-二氯苯溶液,浓度为0.03095ug/ml的N,N-二甲基苯胺溶液,进样量为6 ul时产生的主峰信号约为噪音的3倍;以信噪为在10:1为定量限进行测定,结果表明浓度为0.2054ug/ml的1,2-二氯苯溶液,浓度为0.1548ug/ml的N,N-二甲基苯胺溶液,进样量为6 ul时产生的主峰信号约为噪音的10倍。
2.9【2】方法耐用性
分别测定2.3中的1,2-二氯苯和N,N-二甲基苯胺标准对照溶液放置0、1、2、4、6、8、24、48小时后其峰面积RSD=0.61% 和RSD=1.62%;分别使用南京化学试剂有限公司和上海化学试剂有限公司的无水乙醇配制对照液进行测定,使用DB-624(30m×0.53mm×3.0um) NO:us5342227H和DB-624(30m×0.53mm×3.0um) NO:us7120822H两个批号的色谱柱,测定结果均未有差异;分别设定柱温温度148℃、150℃、152℃,柱流速为3.8ml/min、4.0 ml/min、4.2 ml/min,平衡温度为85℃、90℃、95℃,平衡时间为8min、10 ml/min、12 ml/min,测定1,2-二氯苯和N,N-二甲基苯胺标准对照溶液,其结果都表明无较明显变化。
2.10【2】样品测试
分别称取FLC2009034、FLC2009035、FLC2009036三个批号的样品1.0g于10ml容量瓶中,用无水乙醇定容,将2.2.1的标准对照液和三批样品进行测试,结果显示该三批样品中的1,2-二氯苯和N,N-二甲基苯胺均未检出。
3 讨论
本方法溶剂出峰与两个标准峰能较好分离,空白溶剂不干扰测试;本方法灵敏度高、重现性好、分离效果好,具有很好的线性、准确性和耐用性,本方法能有效准确地控制氟胞嘧啶产品中1,2-二氯苯和N,N-二甲基苯胺两种溶剂残留,是一个实用可靠的检测方法。
参考文献
【1】2011年1月欧洲药品质量委员会出版的Ph.Eur.7.0药典第123-127页,2011年1月美国药典委员会出版的USP34药典第171-182页。
【2】2005年人用药物注册技术要求国际协调会发布的ICHQ2指南和2010年中国医药科技出版社出版的中国药典2010版二部附录194-195页。