分子对接
分子对接的计算方法

分子对接的计算方法分子对接是研究分子之间相互作用的重要方法之一,也是药物设计和发现的重要手段。
本文将介绍分子对接的计算方法,包括分子对接的基本原理、常用的对接算法以及对接评价指标等内容。
一、分子对接的基本原理分子对接是指研究两个分子在特定条件下结合的过程。
在一个分子中,不同原子之间的结合方式是由不同的化学键决定的。
因此,在分子对接中,我们需要考虑来自两个分子之间的多种相互作用,如氢键、电荷相互作用和范德华力等。
分子对接的过程可以分为四步:1)预处理流程;2)构建计算模型;3)执行对接过程;4)结果分析。
在预处理流程中,需要进行双分子的构形搜索,即为每个分子寻找能量最低的构象以用于后面的分子对接计算。
在构建计算模型中,需要将每个分子的三维结构转化为能够进行计算的结构,并将两个分子同时输入计算机。
在执行对接过程中,需要调用分子对接算法,以计算两个分子之间的相互作用,找到最佳的配对方式。
最后,在结果分析中,需要评估分子对接的结果以确定这些分子是否适合结合,以及确定最佳的结合方式。
二、常用的对接算法为了找到最佳的配对方式,许多不同的分子对接算法已被开发出来。
这些算法中的一些常见的方法包括以下内容:1. 基于蒙特卡罗的对接方法基于蒙特卡罗(MC)的对接方法是一种在二维空间中进行随机游走的方法,通过模拟蒙特卡罗过程来寻找最佳的配对方式。
这种方法的优点在于其适合于使用分子动力学模拟技术进行计算,在计算中可以考虑原子或者分子之间的动态变化,更加真实地反映实验情况。
2. 基于分子力学的对接方法基于分子力学的对接方法是一种基于分子动力学模拟的方法。
该方法使用分子动力学技术来计算化学作用过程中的原子或分子的位置和速度变化。
由于该方法考虑了分子内部的相互作用和外部的环境条件对分子结合曲线和内部能量的影响,因此它比其他对接方法更加准确。
3. 基于评分函数的对接方法基于评分函数的对接方法是一种对接评估技术,它借助实验中已经被众所周知的分子结合模型来评估分子结合的力度。
分子对接的概念

分子对接的概念
嘿,大家好啊!今天咱来说说“分子对接”是啥玩意儿。
我给你打个比方哈,就说有一次我玩拼图游戏。
你看,那些拼图小块得找到合适的位置才能拼在一起,形成一个完整的图案。
这分子对接就有点像这个。
分子对接呢,就是让两个分子像拼图块一样凑到一块儿。
比如说一种药和身体里的某个蛋白质分子。
科学家就想让这个药分子和蛋白质分子能准确地对接上。
如果对接得好,这个药就能发挥作用,治好病。
想象一下,这两个分子在一个微观世界里,左晃晃右晃晃,找着那个最合适的位置。
就跟咱玩拼图的时候,这儿试试那儿试试,最后“咔”一下对上了一样。
在现实生活中,科学家们就一直在研究分子对接,想找到最好的办法让那些有用的分子能和该对接的分子完美结合。
这样就能开发出更好的药啊、材料啥的。
所以啊,分子对接就是让两个分子像玩拼图一样找到最合适的结合方式,挺神奇的吧?这就是分子对接的概念啦。
简述分子对接的原理和应用

简述分子对接的原理和应用1. 概述分子对接是一种计算化学方法,用于模拟和预测分子之间的相互作用以及它们在生物体内的相互作用。
分子对接在药物设计、蛋白质研究等领域中有广泛的应用。
2. 分子对接的原理分子对接的原理基于分子之间的相互吸引力和排斥力。
分子对接通过将配体(小分子)与靶标位点(蛋白质)进行模拟,预测它们之间的最佳结合模式。
3. 分子对接的步骤分子对接通常包括以下步骤:(1) 数据准备在分子对接之前,需要准备配体和靶标的三维结构信息,通常是通过实验测定或计算方法得到的。
(2) 靶标位点的预测如果靶标的结构未知,可以通过蛋白质比对、结构预测等方法获取其三维结构。
(3) 配体库的筛选根据疾病相关的生物通路和目标蛋白的特异性,从配体库中选出潜在的配体候选物。
(4) 对接模拟通过使用分子对接软件,将配体与靶标进行模拟,预测它们之间的最佳结合模式。
这通常涉及到搜索和评分两个主要的步骤。
(5) 结果分析根据对接模拟的结果,评估配体与靶标之间的相互作用力,并从中选择具有潜在生物活性的化合物。
4. 分子对接的应用分子对接在药物设计、蛋白质研究等领域中具有广泛的应用。
(1) 药物设计分子对接可以帮助药物设计师预测和优化化合物与靶标的结合模式,从而提高药物的活性和选择性,并减少不必要的实验。
这有助于加速药物研发的过程。
(2) 蛋白质研究分子对接可以用于研究蛋白质和其他生物大分子之间的相互作用,从而揭示其功能和机制。
这对于理解生物过程以及疾病的发生和发展非常重要。
(3) 农药设计分子对接可以应用于农药设计,通过预测农药与害虫靶标之间的结合模式,设计新的、高效的农药,以提高农作物的产量和质量。
(4) 材料科学分子对接还可以用于材料科学领域,通过预测分子在材料表面的结合模式,优化材料的性能,如吸附性、催化活性等。
5. 结论分子对接作为一种重要的计算化学方法,在药物设计、蛋白质研究等领域中有广泛的应用。
它通过模拟分子之间的相互作用,帮助研究者预测和优化化合物与靶标之间的结合模式,并加速药物研发和生物研究的过程。
分子对接 分子生成模型

分子对接分子生成模型
分子对接是一种重要的计算方法,用于预测分子之间的结合方式和力学性质。
通过分子对接,我们可以模拟和研究分子之间的相互作用,从而为药物设计、酶催化机理等领域提供重要的理论指导。
分子对接的过程可以分为三个主要步骤:准备工作、搜索和评价。
在准备工作中,我们需要获得目标分子和配体分子的结构信息,并进行预处理,如去除水分子和离子等。
接下来,我们需要为目标分子和配体分子分别建立力场参数,以便在模拟中准确描述它们之间的相互作用。
搜索是分子对接过程的核心步骤,主要通过搜索算法来探索不同的结合方式。
常见的搜索算法包括基于格点的搜索算法、模拟退火算法、遗传算法等。
在搜索过程中,我们会对目标分子和配体分子的柔性进行考虑,以尽可能地找到最优的结合方式。
评价是对搜索得到的结合方式进行打分和筛选的过程。
通过计算得到的结合能、分子间相互作用能等指标,我们可以评估不同结合方式的稳定性和亲和力。
同时,我们还可以通过分析结合口袋的形状和特征来评估结合方式的合理性。
分子对接模型的建立是分子对接研究的重要一环。
通过对已知结合配体和目标分子的结构信息进行统计分析和机器学习,我们可以建立分子对接模型,用于预测未知分子的结合方式和亲和力。
常见的
分子对接模型包括基于物理原理的模型、基于机器学习的模型等。
分子对接是一种重要的计算方法,通过模拟和研究分子之间的相互作用,为药物设计等领域提供重要的理论支持。
分子对接模型的建立可以进一步提高对未知分子结合方式的预测准确性,为药物研发和生物研究提供有力的工具。
分子对接全

蛋白质二级结构的主要形式
• -螺旋 ( -helix ) • -折叠 ( -pleated sheet ) • -转角 ( -turn ) • 无规卷曲 ( random coil )
-螺旋
-折叠
-转角和无规卷曲
-转角
无规卷曲是用来阐述没有确定规律性的那部 分肽链结构。
❖ 定量指标,需要结合分子动力学进一步评价
AutoGrid 格点中相关能量的计算
AutoDock 构象搜索及评价
❖ 免费软件/
AutoDock分子对接的流程:
1.围绕受体活性位点的氨基酸残基形成一个盒 子(box),并划分成格点; 2.用配体不同类型的原子作为探针(probe)进 行扫描,计算格点能量; 3.对配体在box范围内进行构象搜索; 4.根据配体的不同构象、方向、位置及能量进 行评分,最后对结果进行排序。
蛋白质分子中各亚基的空间排布及亚基接 触部位的布局和相互作用,称为蛋白质的四级 结构。
亚基之间的结合力主要是疏水作用,其次 是氢键和 结 构
从一级结构到四级结构
血红蛋白
二、酶及其抑制剂
酶是由活细胞产生的对其特异的底物 起高效催化作用的蛋白质。
酶的分子组成
❖ 单纯酶(simple enzyme):仅由氨基酸残基构成 ❖ 结合酶(conjugated enzyme)
苏氨酸 threonine Thr T 5.60
3. 酸性氨基酸 4. 碱性氨基酸
天冬氨酸 aspartic acid Asp D 2.97 谷氨酸 glutamic acid Glu E 3.22
赖氨酸
lysine
Lys K 9.74
精氨酸 arginine Arg R 10.76
分子对接
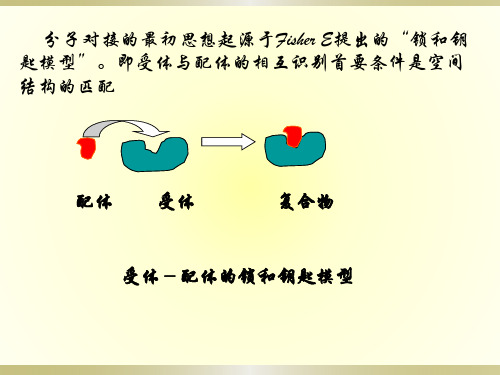
分子对接的基本原理
药物与受体的结合强度取决于结合的自由能变化
∆G结合 = ∆H结合 - T∆S结合 = -RT ln Ki
大部分的分子对接法忽略了全部的熵效应,而在焓 效应也只考虑配体与受体的相互作用能,即:
Einteraction= Evdw + Eelectrostatic + Eh-bond
(2)基于几何哈希技术“geometric hashing”的方法
第一部分中,几何哈希表从被对接的一个配体或一系 列配体中构建 。哈希矩阵含有配体名字和能调整配体在空 间方向的参考框架。
第二部分即识别阶段,蛋白质的特征用来识别哈希矩阵, 每一次匹配表示蛋白质的特征与哈希矩阵中已定义好方位的 配体相匹配,具有大量匹配信息的哈希矩阵代表着具有几个 吻合特征的配体和方位
分子对接的基本方法
(一) 刚性的分子对接方法
这种方法是最初的分子对接的方法,在对接中,小 分子和蛋白质两种都保持刚性。
(1)基于最大团搜索的方法 (Clique-Search Based Approaches)
对接两个刚性分子可以理解为分子在空间的匹配问 题,这种匹配可以是一种形状上的互补或相互作用。 如氢键受体与氢键给体的互补。搜索在三维空间中有 效的条件下的最大匹配
(3) 遗传算法和进化规划
遗传算法开始应用到分子对接技术,其特点为 :
第一步,一个称为染色体的线性表示符能够描述构型的所有 自由度,找到这个染色体描述符是算法中最困难的一步。第 二步,确定一个一个类似如打分函数的目标函数。
著名的GOLD软件包括了这种算法
(4)基于分子模拟的方法
模拟退火的方法,Autodock程序就采用了这种方法
(二)柔性对接的方法
分子对接简要介绍
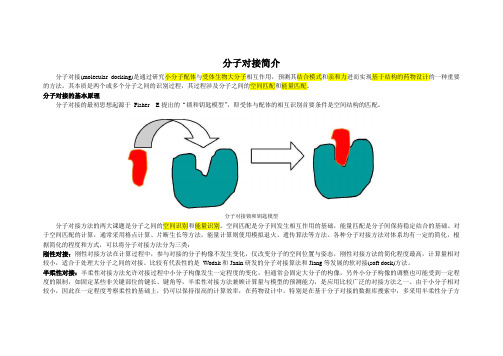
分子对接简介分子对接(molecular docking)是通过研究小分子配体与受体生物大分子相互作用,预测其结合模式和亲和力进而实现基于结构的药物设计的一种重要的方法。
其本质是两个或多个分子之间的识别过程,其过程涉及分子之间的空间匹配和能量匹配。
分子对接的基本原理分子对接的最初思想起源于Fisher E提出的“锁和钥匙模型”,即受体与配体的相互识别首要条件是空间结构的匹配。
分子对接锁和钥匙模型分子对接方法的两大课题是分子之间的空间识别和能量识别。
空间匹配是分子间发生相互作用的基础,能量匹配是分子间保持稳定结合的基础。
对于空间匹配的计算,通常采用格点计算、片断生长等方法,能量计算则使用模拟退火、遗传算法等方法。
各种分子对接方法对体系均有一定的简化,根据简化的程度和方式,可以将分子对接方法分为三类:刚性对接:刚性对接方法在计算过程中,参与对接的分子构像不发生变化,仅改变分子的空间位置与姿态,刚性对接方法的简化程度最高,计算量相对较小,适合于处理大分子之间的对接。
比较有代表性的是Wodak和Janin研发的分子对接算法和Jiang等发展的软对接(soft dock)方法。
半柔性对接:半柔性对接方法允许对接过程中小分子构像发生一定程度的变化,但通常会固定大分子的构像,另外小分子构像的调整也可能受到一定程度的限制,如固定某些非关键部位的键长、键角等,半柔性对接方法兼顾计算量与模型的预测能力,是应用比较广泛的对接方法之一。
由于小分子相对较小,因此在一定程度考察柔性的基础上,仍可以保持很高的计算效率,在药物设计中,特别是在基于分子对接的数据库搜索中,多采用半柔性分子方法。
其代表性软件是DOCK和AutoDock。
柔性对接:柔性对接方法在对接过程中允许研究体系的构像发生自由变化,由于变量随着体系的原子数呈几何级数增长,因此柔性对接方法的计算量非常大,消耗计算机时很多,适合精确考察分子间识别情况。
其中比较有代表性的方法有Accelrys 公司发展的基于分子力学和分子动力学的分子对接方法及Affinity 软件。
分子对接

分子对接(molecular docking)使依据配体与受体作用的“锁-钥原理”(lock and key principle),模拟小分子配体与受体生物大分子相互作用。
配体与受体相互作用是分子识别的过程,主要包括静电作用、氢键作用、疏水作用、范德华作用等。
通过计算,可以预测两者间的结合模式和亲和力,从而进行药物的虚拟筛选。
分子对接首先产生一个填充受体分子表面的口袋或凹槽的球集,然后生成一系列假定的结合位点。
依据受体表面的这些结合点与配体分子的距离匹配原则,将配体分子投映到受体分子表面,来计算其结合的模式和亲和力,并对计算结果进行打分,评判配体与受体的结合程度。
2 分子对接的原理2. 1 分子对接的一般原理分子对接是将已知三维结构数据库中的分子逐一放在靶标分子的活性位点处。
通过不断优化受体化合物的位置、构象、分子内部可旋转键的二面角和受体的氨基酸残基侧链和骨架, 寻找受体小分子化合物与靶标大分子作用的最佳构象, 并预测其结合模式、亲和力和通过打分函数挑选出接近天然构象的与受体亲和力最佳的配体的一种理论模拟分子间作用的方法。
2. 2 分子对接的互补性影响复合物分子稳定性的主要因素是疏水作用和键合力大小, Audie 等[ 2] 建立了一种新的经验自由能公式, 该方法能够预测蛋白质2蛋白质键合力大小, 准确率达到89% 。
而影响键合力的因素有作用位点空间位的互补、静电相互作用和氢键等, 且溶解熵对稳定受体2配体复合物起着重要的作用[ 3] , 所以分子对接的过程主要包括分子间的空间互补和电学性质互补。
空间互补是分子间发生相互作用的基础, 能量互补是分子间保持稳定结合的基础。
3 分子对接的种类分子对接的种类主要包括:(1)刚体对接:指在对接过程中,研究体系(受体和配体)的构象不发生变化。
适合考察比较大的体系,如蛋白质和蛋白质间以及蛋白质和核酸之间的对接。
(2)半柔性对接:指在对接过程中,研究体系尤其是配体的构象允许在一定的范围内变化。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
2.分子对接的原理
• 2.1 分子对接的一般原理
• 分子对接是将已知三维结构数据库中的分子逐一 放在靶标分子的活性位点处。通过不断优化受体 化合物的位置、构象、分子内部可旋转键的二面 角和受体的氨基酸残基侧链和骨架,寻找受体小分 子化合物与靶标大分子作用的最佳构象,并预测其 结合模式、亲和力和通过打分函数挑选出接近天 然构象的与受体亲和力最佳的配体的一种理论模 拟分子间作用的物分子稳定性的主要因素是疏水作用和 键合力大小。 • 而影响键合力的因素有作用位点空间位的互补、 静电相互作用和氢键等,且溶解熵对稳定受体-配体 复合物起着重要的作用。 • 所以分子对接的过程主要包括分子间的空间互补 和电学性质互补。 • 空间互补是分子间发生相互作用的基础,能量互补 是分子间保持稳定结合的基础。
• 3.3柔性对接
• 在对接过程中,配体和受体的构象是允许发生变化 的。一般用于精确考虑分子间的识别情况。 • 分子的柔性主要来自于可旋转健的旋转。这种变 化包括三个平动自由度、三个转动自由度以及配 体分子的部分二面角的变化。
4.主要分子对接软件
• 如果要进行分子对接试验,就必须先对所要对接的 分子进行结构模拟。当分子模型确立后,就可以利 用分子模拟软件在计算机上进行一系列实验。主 要软件有:
6.展望
• 分子对接方法的优势在于各种化合物数据库中的 分子均是已知化合物,且相当大一部份可以通过购 买得到或根据已知的合成路线合成,可以较快地进 行后续的药理测试。目前有许多的商用数据库,为 药物开发提供了一个比较好的工具。近年来,计算 机技术的发展、靶酶晶体结构数据和算法数量的 快速增长和商用小分子数据库的不断更新,使得分 子对接在药物设计中取得了巨大成功。
Stoddard等用二元对接方法成功地对麦芽糖和蛋白 质进行了对接。对接时,他们把配体和受体主链结 构当作刚性处理。
• 3.2半柔性对接
• 在对接过程中,研究体系尤其指配体的构象允许在 一定的范围内变化,但大分子是刚性的,构象不变。
比如蛋白质分子与配体小分子的作用。进行半柔 性对接时,一般使用快速的对接软件-FIexX,因为它 允许配体是柔性,而受体是刚性的。
• 2.2.2能量匹配
• 配体与受体能量匹配包括静电作用、氢键作用、 范德华相互作用和疏水作用等。 • 静电作用、氢键作用和范德华力控制药物-受体结 合。 • 疏水作用是药物-受体结合的驱动力。
3.分子对接方法分类
• 3.1刚体对接
• 3.2半柔性对接 • 3.3柔性对接
• 3.1刚体对接
• 在对接过程中,研究体系(主体与客体)的构象不发 生变化。适合考察结构比较大的体系,如蛋白质和 蛋白质间以及蛋白质和核酸等大分子间的对接。
参考文献
• 段爱霞,陈 晶,刘宏德,刘秀辉,卢小泉 分子对接方 法的应用与发展 分析科学学报 第25卷第4期 2009年8月 • 朱志远;张燕; 李征; 李金恒; 芮建中; 袁靖 受体蛋白与药物分子对接的研究进展 中国临床药理学与治疗学 2009-11-26期刊 • 赵丽琴;肖军海;李松 分子对接在基于结构药物设 计中的应用 生物物理学报 2002-09-30期刊
(1)对于已知生物活性但作用机制尚不明确的候选 药物,如天然产物或化学合成物,分子反向对接 可以帮助确定这些化合物潜在结合靶蛋白,从而 为作用机制的解析提供有益思路。
(2)对于作用机制明确的已知药物或药物先导物, 通过反向对接搜寻可以提供化合物次级结合蛋白 信息,这无疑对于“老药新用”的发展具有重要 作用。
• • • • •
Dock(第一个分子对接软件) AutoDock Hyperchem MOE(Molecular Operating Environment) InsightII 2005
5.分子对接的新应用
• 除了基于相同受体进行的小分子配体数据库搜寻 计算的一般意义上的分子对接方法。 • 近几年,一种新的配体-受体反向对接方法被提出 ,用于小分子配体潜在结合蛋白分子的搜寻。反 向对接及蛋白数据库搜寻在药物研究中具有诸多 潜在应用。其两大优点为:
分子对接
Molecular Docking
组员: 应用化学1201班1号至11号 教师 :张老师
Content
• • • • • • 1.分子对接概念 2.分子对接的原理 3.分子对接方法分类 4.主要分子对接软件 5.分子对接的新应用 6.展望 参考文献
1.分子对接概念
• 分子对接 (Molecular Docking)已成为药物设 计方法中比较成熟的直接药物设计方法。 • 从已知结构的受体(靶蛋白或活性位点)和配 体出发,通过化学计量学方法模拟分子的几 何结构和分子间作用力来进行分子间相互 作用识别并预测受体-配体复合物结构的方 法称为分子对接。
• 2.2.1空间互补
• 目前,最常见的研究受体大分子结合部位三维结构 的方法是X-单晶衍射(蛋白质晶体学),其次是二维 核磁共振和多维核磁研究液相结构,电镜三维重组 、电子衍射、中子衍射和各种频谱学方法研究生 物高分子的空间结构。尽管受体大分子的三维结 构能够通过以上方法得到,但当不同的配体靠近受 体时,往往会因为分子的柔性而导致受体分子的结 构发生变化。