Organic Syntheses, Coll. Vol. 3, p.207 (1955); Vol. 21, p.18 (1941).
DBA的合成

Organic Syntheses, CV 2, 167DIBENZALACETONE[3-Pentanone, 1,5-diphenyl-]Submitted by Charles R. Conard and Morris A. Dolliver.Checked by H. Lohse and C. R. Noller.1. ProcedureA cooled solution of 100 g. of sodium hydroxide in 1 l. of water and 800 cc. of alcohol (Note 1) is placed in a 2-l. wide-mouthed glass jar which is surrounded with water and fitted with a mechanical stirrer. The solution is kept at about 20–25° and stirred vigorously (Note 2) while one-half of a mixture of 106 g. (1 mole) of benzaldehyde and 29 g. (0.5 mole) of acetone is added (Note 3). In about two or three minutes a yellow cloud forms which soon becomes a flocculent precipitate. After fifteen minutes the rest of the mixed reagents is added, and the container is rinsed with a little alcohol which is added to the mixture. Vigorous stirring is continued for one-half hour longer, and the mush is then filtered with suction on a large Büchner funnel. The product is thoroughly washed with distilled water (Note 4) and then dried at room temperature to constant weight. The yield is 105–110 g. (90–94 per cent of the theoretical amount) (Note 5) of a product which melts at 104–107°.The crude dibenzalacetone may be recrystallized from hot ethyl acetate, using 100 cc. of solvent for each 40 g. of material. The recovery in this purification is about 80 per cent; the purified product melts at 110–111°.2. Notes1. Sufficient alcohol is used to dissolve the benzaldehyde rapidly and to retain the benzalacetone in solution until it has had time to react with the second molecule of aldehyde. Lower concentrations of base slow up the formation of the dibenzalacetone and thus favor side reactions which yield a sticky product. Higher concentrations of base give added difficulty in washing. These concentrations were suggested by, and are approximately the same as, those used in the preparation of benzalacetophenone described in Org. Syn. Coll. V ol. I, 1941, 78.2. Only temperatures between 20 and 25°were tried; it was assumed that a change of temperature would have the same effect that it has in the preparation of benzalacetophenone mentioned above.Stirring is essential, as it makes considerable difference in the uniformity of the product.3. The benzaldehyde was u.s.p. quality which had been washed with sodium carbonate solution and distilled. Commercial c.p. acetone was used. The theoretical quantities are used, since an excess of benzaldehyde results in a sticky product while an excess of acetone favors the production of benzalacetone. The mixture is prepared before addition in order to ensure additions of equivalent quantities.4. Since the product is practically insoluble in water, large amounts can be used in the washing. Sodium compounds are probably the chief impurities. The dried product contains somesodium carbonate which results from the failure to remove the sodium hydroxide completely. There remain also the impurities insoluble in water. However, the product is pure enough for use in most reactions.5. If the mush is allowed to stand several hours, chilled, and filtered cold, a slightly larger yield is obtained, but this is not worth while. The filtrate may be used as a medium for a second run in which about 93 per cent of the theoretical yield is obtained. The melting point of the second product is slightly lower.3. DiscussionDibenzalacetone has been prepared by condensing benzaldehyde with acetone using as condensing agents dry hydrogen chloride,1 10 per cent sodium hydroxide solution,2 and glacial acetic acid with sulfuric acid.3It has also been obtained by condensing benzalacetone with benzaldehyde in the presence of dilute sodium hydroxide.4Straus and Ecker5were the first to record the use of ethyl acetate for crystallization.References and NotesClaisen and Claparède, Ber. 14, 350 (1881).Schmidt, ibid. 14, 1460 (1881); Claisen, ibid. 14, 2470 (1881); Straus and Caspari. ibid. 40, 2698 (1907).Claisen and Claparède, ibid. 14, 2460 (1881).Claisen and Ponder, Ann. 223, 141 (1884).Straus and Ecker, Ber. 39, 2988 (1906).AppendixCompounds Referenced (Chemical Abstracts Registry Number)alcohol (64-17-5)sulfuric acid (7664-93-9)hydrogen chloride (7647-01-0)acetic acid (64-19-7)ethyl acetate (141-78-6)sodium hydroxide (1310-73-2)sodium carbonate (497-19-8)Benzalacetophenone (94-41-7)benzaldehyde (100-52-7)acetone (67-64-1)Benzalacetone (122-57-6)Dibenzalacetone (35225-79-7)3-Pentanone, 1,5-diphenyl-。
库尔提斯重排反应

库尔提斯重排反应[编辑](重定向自Curtius重排反应)库尔提斯重排反应(Curtius重排反应)是一个重排反应,首先由库尔提斯(Theodor Curtius)发现,反应中酰基叠氮重排生成异氰酸酯。
[1][2]关于此反应的综述参见:[3][4]。
产物可与一系列亲核试剂反应:与水作用水解得到胺;[5]与苯甲醇反应生成带有苄氧羰基保护基(Cbz)的胺类;[6]与叔丁醇作用生成带有叔丁氧羰基保护基(Boc)的胺类,用作有机合成中的重要中间体。
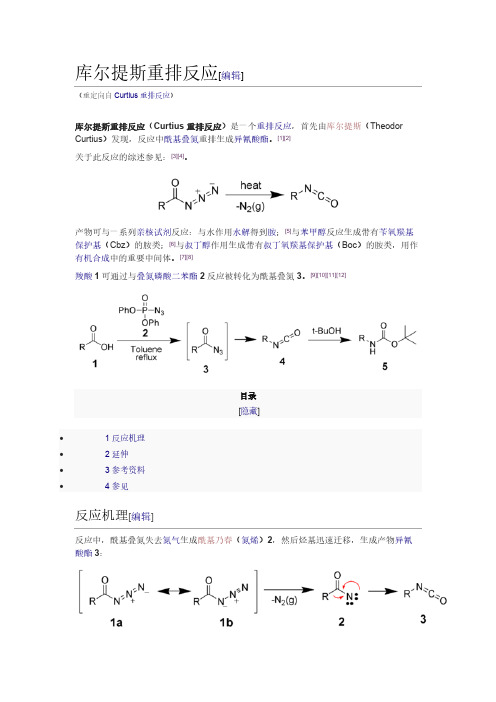
[7][8]羧酸1可通过与叠氮磷酸二苯酯2反应被转化为酰基叠氮3。
[9][10][11][12]目录[隐藏]∙ 1 反应机理∙ 2 延伸∙ 3 参考资料∙ 4 参见反应机理[编辑]反应中,酰基叠氮失去氮气生成酰基乃春(氮烯)2,然后烃基迅速迁移,生成产物异氰酸酯3:延伸[编辑]在Curtius重排反应的基础上,Darapasky递降反应(A. Darapsky, 1936)以α-氰基酯为原料,通过重排反应生成氨基酸。
[13]参考资料[编辑]1. ^ Curtius, T. Ber.1890, 23, 3023.2. ^ Curtius, T. J. Prakt. Chem.1894, 50, 275.3. ^ Smith, P. A. S. Org. React.1946, 3, 337-449. (Review)4. ^ Scriven, E. F.; Turnbull, K.; Chem. Rev.1988, 88, 297-368. Review5. ^ Kaiser, C.; Weinstock, J. Organic Syntheses, Coll. Vol. 6, p.910 (1988); Vol. 51,p.48 (1971).Article6. ^ Ende, D. J. a.; DeVries, K. M.; Clifford, P. J.; Brenek, S. J. Org. Proc. Res.Dev.1998, 2, 382-392.7. ^ Lebel, H.; Leogane, O.; Org. Lett.2005, 7(19),4107-4110. doi:10.1021/ol051428b8. ^ Shioiri, T.; Yamada, S. Organic Syntheses, Coll. Vol. 7, p.206 (1990); Vol. 62,p.187 (1984).Article9. ^ Shioiri, T.; Ninomiya, K.; Yamada, S. J. Am. Chem. Soc.1972, 94,6203-6205.doi:10.1021/ja00772a05210. ^ Ninomiya, K.; Shioiri, T.; Yamada, S. Tetrahedron1974, 30, 2151-2157.11. ^ Wolff, O.; Waldvogel, S. R. Synthesis2004, 1303-1305.12. ^ Jessup, P. J.; Petty, C. B.; Roos, J.; Overman, L. E. Organic Syntheses, Coll.Vol. 6, p.95 (1988); Vol. 59, p.1 (1979). Article13. ^/reactions/RXN051.htm(重定向自贝克曼重排)贝克曼重排反应(Beckmann rearrangement)是一个由酸催化的重排反应,反应物肟在酸的催化作用下重排为酰胺。
钯碳 水合肼 还原 硝基

Organic Syntheses, Coll. Vol. 5, p.30 (1973); Vol. 40, p.5 (1960).2-AMINOFLUORENE[2-Flurenylamine]Submitted by P. M. G. Bavin1Checked by John C. Sheehan and Roger E. Chandler..1. ProcedureIn a 2-l. three-necked round-bottomed flask, equipped with a mechanical stirrer (Note 1), reflux condenser, and dropping funnel, are placed 30 g. of pure 2-nitrofluorene, m.p. 157° [Org. Syntheses, Coll. Vol. 2, 447 (1943)], and 250 ml. of 95% ethanol. After warming to 50° on a steam bath, 0.1 g. of palladized charcoal catalyst (previously moistened with alcohol) is added (Note 2) and the stirrer is started. About 15 ml. of hydrazine hydrate is added from the dropping funnel during 30 minutes (Note 3). At this point an additional 0.1 g. of catalyst (previously moistened with alcohol) is added and the mixture is heated until the alcohol refluxes gently. After 1 hour the nitrofluorene has dissolved completely and the supernatant liquor is almost colorless.The catalyst is removed by filtration with gentle suction through a thin layer of Celite (Note 4). The flask is rinsed with 30 ml. of hot alcohol which is then used to wash the catalyst and Celite. The combined filtrates are concentrated under reduced pressure to about 50 ml. (Note 5) and then heated to boiling at atmospheric pressure. When 250 ml. of hot water is added slowly, 2-aminofluorene is precipitated as a colorless, crystalline powder. After cooling in an ice bath, the 2-aminofluorene is collected, washed with water, and dried in the dark in a vacuum desiccator. The product melts at 127.8–128.8° (Note 6) and amounts to 24–25 g. (93–96%).2. Notes1. If the stirring is omitted, the nitrofluorene takes longer to dissolve.2. A suitable catalyst is 10% palladium-on-charcoal, such as is supplied by Baker and Company, Inc., 113 Astor Street, Newark 5, New Jersey.3. The reaction is exothermic, and too rapid addition of the hydrazine may cause the mixture to foam out of the condenser.4. Caution! The catalyst is often pyrophoric and should be kept moistened with alcohol. Celite is a diatomaceous earth filter aid.5. A rotary evaporator is very convenient for the concentration since some of the amine invariably crystallizes toward the end.6. The melting point is that reported in Organic Syntheses, Coll. Vol. 2, 448 (1943), for a recrystallized sample.3. DiscussionThe preparation of 2-aminofluorene reported previously in Organic Syntheses[Coll. Vol. 2, 448 (1943)] we based on the method of Diels.2The present procedure illustrates a general method for the reduction of aromatic nitro compounds to aromatic amines using hydrazine and a hydrogenation catalyst such as palladium, platinum, nickel, iron, or rethenium. The literature on this procedure up to 1963 has been reviewed.3 In many instances the catalytic hydrazine reductions give yields of amine equal to or better than those obtained by directcatalytic hydrogenation or other reduction methods. Both the apparatus and the procedure are simple. Under appropriate conditions the method may be used for the dehalogenation of aliphatic and aromatic halides,3 a reaction for which palladium appears to be a specific catalyst. The method has also been used for the reduction of azobenzene and azoxybenzene to hydrazobenzene (80–90%),4 as well as for the synthesis of steroid aziridines by reduction of mesylate esters by vicinal azido alcohols (using Raney nickel).5References and Notes1.National Research Council of Canada Post-doctorate Fellow, 1954-56, at the University ofOttawa, Ottawa, Ontario.2.O. Diels, Ber., 34, 1758 (1901).3. A. Furst, R. C. Berlo, and S. Hooton, Chem. Rev., 65, 51 (1965).4.P. M. G. Bavin, Can. J. Chem., 36, 238 (1958).5.K. Ponsold, Ber., 97, 3524 (1964).AppendixChemical Abstracts Nomenclature (Collective Index Number);(Registry Number)2-Flurenylaminepalladized charcoal catalystpalladium-on-charcoalethanol (64-17-5)iron (7439-89-6)platinum (7440-06-4)nickel,Raney nickel (7440-02-0)palladium (7440-05-3)hydrazine hydrate (7803-57-8)hydrazine (302-01-2)Azoxybenzene (495-48-7)Azobenzene (103-33-3)2-Nitrofluorene (607-57-8)2-Aminofluorene(153-78-6)Nitrofluoreneretheniumhydrazobenzene (122-66-7) Copyright © 1921-2005, Organic Syntheses, Inc. All Rights Reserved。
关于羧酸酯还原为醇的原理

TBSO OH NMe2
Organic Syntheses,Coll.Vol.10,p.442;Vol.78,p.160
Work up: The reaction mixture is stirred for 15 min, diluted with ether(100 mL), and quenched by dropwise addition of water (9 mL). The resulting gray suspension is allowed to reach RT, and the mixture is stirred vigorously for an additional 60 min.The mixture is transferred to a 1.0-L Erlenmeyer flask and diluted with ether(350 mL).
NaBH4还原
NaBH4 H3CO2C N CO2CH3 HO N OH
Liebigs Ann.lRecueil 1997,707-720. 此法操作较为简单,安全.由于NaBH4的还原性不够强,因此此类反应一 般需要回流过夜.而且,反应初始阶段不要去加热,而是在室温下搅拌数小 时后再缓缓加热至回流,否则极易冲料.
关于羧酸酯还原为醇 的原理及操作
• 1.金属钠和醇为还原剂(BouveaultBlanc反应) • 2.金属氢化物为还原剂
金属钠和醇为还原剂
• 本反应是将羧酸酯用 金属钠和醇直接还原 生成相应的伯醇,主 要用于高级脂肪羧酸 酯的还原.
O n-C11H23 OEt Na,EtOH toluene n-C11H23 OH
NaBH4-ZnCl2还原
EtOOC OTIPS COOEt Zn(BH4)2 THF EtOOC OTIPS OH
库尔提斯重排反应-推荐下载
在一个研究中[5],研究者使用电脑模拟丙酮肟在贝克曼溶剂中的重排反应,并考虑到了溶 剂分子和取代物的影响。模拟表明,有三个乙酸分子和一个质子(以氧鎓的形式存在)参 与了反应。形成亚胺中间体后(σ 配合物),甲基通过协同反应迁徙到氮上,并推走羟基。 羟基中氧原子受到三个乙酸分子的稳定。接下来,一分子水进攻亲电的碳原子,其中一个 氢原子被一个乙酸接收,生成的中间体为 N-甲基乙酰氨酸,其中氧原子为四配位。最后异 构化形成稳定的产物酰胺。
p.48 (1971).Article 6. ^ Ende, D. J. a.; DeVries, K. M.; Clifford, P. J.; Brenek, S. J. Org. Proc. Res.
Dev. 1998, 2, 382-392. 7. ^ Lebel, H.; Leogane, O.; Org. Lett. 2005, 7(19), 4107-
延伸[编辑]
在 Curtius 重排反应的基础上,Darapasky 递降反应(A. Darapsky, 1936)以 α-氰基酯 为原料,通过重排反应生成氨基酸。[13]
参考资料[编辑]
1. ^ Curtius, T. Ber. 1890, 23, 3023. 2. ^ Curtius, T. J. Prakt. Chem. 1894, 50, 275. 3. ^ Smith, P. A. S. Org. React. 1946, 3, 337-449. (Review) 4. ^ Scriven, E. F.; Turnbull, K.; Chem. Rev. 1988, 88, 297-368. Review 5. ^ Kaiser, C.; Weinstock, J. Organic Syntheses, Coll. Vol. 6, p.910 (1988); Vol. 51,
Organic Syntheses, Coll. Vol. 4, p.840 (1963); Vol. 36, p.79 (1956).

Organic Syntheses, Coll. Vol. 4, p.840 (1963); Vol. 36, p.79 (1956).SEBACOIN[Cyclodecanone, 2-hydroxy-]Submitted by Norman L. Allinger1Checked by N. J. Leonard, J. C. Little, and F. H. Owens.1. ProcedureThe apparatus2 consists of a 3-l. three-necked round-bottomed creased flask, with standard ball joints and an indented cone-shaped bottom (Note 1), which is heated by means of an electric mantle and is equipped with a high-speed stirrer of stainless steel driven by a 10,000 r.p.m. motor (Note 2). One side neck is fitted with a bulb-type air-cooled condenser (Note 3), on top of which fits a 1-l. pressure-equalizing Hershberg dropping funnel (Note 4). The top of the dropping funnel is connected in turn to a U-tube containing a 1-cm. head of mercury. The entire apparatus is securely fastened to a sturdy support.In the flask is placed 900 ml. of xylene(Note 5), and a slow stream of purified nitrogen(Note 6) is passed through the system from which the dropping funnel has been temporarily removed. The stirrer is run at slow speed, and the solvent is brought to a gentle reflux. The air stream cooling the condenser is shut off, the mercury valve is disconnected from the condenser, and a few milliliters of solvent is allowed to distil out the top of the condenser (Note 7). The dropping funnel (Note 8), containing a solution of 115 g. (0.50 mole) of dimethyl sebacate(Note 9) and 500 ml. of xylene(Note 5) is then inserted between the top of the condenser and the mercury valve. The air to the condenser is then turned on, and the electric mantle is turned off. The solvent is allowed to cool below its boiling point, and the stirrer is gradually brought to a stop. Throughout these operations the nitrogen flow is adjusted to keep air out of the system. Through a side neck is then added 50.6 g. (2.20 g. atoms) of crust-free sodium metal cut into lumps of convenient size. The side neck is closed, and the stirrer and the heater are turned on. The sodium is dispersed by stirring at about 6000–8000 r.p.m. for 10 minutes, and, with continued heating and stirring at a rate somewhat slower (to give suitable mixing), the dropwise addition of the ester solution is begun at such a rate as to be complete in about 24 hours (Note 10) and (Note 11).Heating and stirring are continued for 1 hour after the addition is completed. The stirrer is then slowed, heating is stopped, the heater is removed, and the reaction flask is allowed to cool for about 15 minutes (Note 12). The reaction flask is then cooled in a water bath, and is finally thoroughly cooled in an ice bath. A solution of 140 ml. of glacial acetic acid in an equal volume of xylene is then added dropwise during about 30 minutes with continued cooling and stirring (Note 13). After addition of 450 ml. of water, stirring is stopped, the nitrogen is turned off, and the flask is disconnected from the apparatus. The two-phase mixture is filtered through a large Büchner funnel with suction to remove a small amount of gum, and the filtrate is then poured into a 3-l. separatory funnel. The aqueous phase is drawn off, extracted with 100 ml. of xylene, and is discarded. The xylene phases are washed in series with 100 ml. of water, and are combined and dried with 10 g. of anhydrous magnesium sulfate. The solution is filtered into a 3-l. round-bottomed flask, and the bulk of the xylene is distilled with the aid ofan aspirator (Note 14) and (Note 15). The residue is transferred to a smaller flask and is distilled through a 2-ft. Vigreux column, the fraction boiling at 134–138°/14 mm. or 124–128°/9 mm. being collected as a yellowish liquid weighing 57–63 g. (67–74%). This material solidifies on standing and is sufficiently pure for most purposes (Note 16) and (Note 18). For further purification it may be crystallized from 150 ml. of pentane by cooling to −10° in an ice-salt bath for several hours. The mixture is filtered, and the crystals are washed with 50 ml. of pentane which has been cooled to −80°. The pure product thus obtained is a white granular crystalline solid, m.p. 38–39°, weighing 53–56 g. (63–66%) (Note 17) and (Note 18).2. Notes1. A flask having this shape gives the most efficient mixing.22. A one-fourth-horsepower motor is adequate. A suitable motor is manufactured by Bodine Electric Company, Chicago, Illinois.3. The use of a water-cooled glass condenser is not recommended since it might accidentally be broken and thereby cause water to flow into the flask. A metal water-cooled condenser has also been used and is satisfactory.4. Adapted from that described in Org. Syntheses, Coll. Vol. 2, 129 (1943).5. The xylene used was purified by heating under reflux with sodium overnight and then distilling, b.p. 137–142°.6. Linde high-purity dry nitrogen was used without further treatment.7. The fumes may be taken off by attaching an aspirator. This procedure assures removal of all moisture from the system.8. The funnel is dried before use with a flame and is then closed with a drying tube and allowed to cool.9. The ester used was Eastman Kodak Company technical grade shaken with sodium carbonate, dried and distilled. The ester boils at 158–160°/11 mm.10. The reaction time can be lengthened considerably without effect. If, however, the time is shortened appreciably, the yield may be markedly lowered.11. Initially the reaction may take on various colors, red, purple, etc., but after a short time a dull gray-brown color appears, which is gradually replaced by a yellow-brown or olive-drab color.12. It is important that the reaction mixture be kept out of contact with the air until it has been acidified.13. When sufficient acetic acid has been added, the dark color of the reaction mixture is replaced by a white color, and the mixture is often quite thick. More acetic acid is not harmful.14. Nitrogen is led through the capillary during the distillations.15. The xylene thus recovered is purified (Note 5) and is used in the next preparation.16. This material slowly decomposes upon standing. It may be stored for at least several months with only slight decomposition if it is kept under nitrogen in the dark and at 0°. The compound appears to be stable when pure.17. Homologs having a ring containing 10 to 18 carbons have been prepared in an analogous manner in yields from 46 to 85%.318. This preparation has also been carried out on a 1.0 mole scale by the checkers, using a 5-l. creased flask. Comparable yields are obtainable.3. DiscussionSebacoin has been prepared by the cyclization of methyl or ethyl sebacate with sodium metal.3,4,5,6,7,8 This preparation is referenced from:z Org. Syn. Coll. Vol. 4, 216z Org. Syn. Coll. Vol. 4, 218z Org. Syn. Coll. Vol. 4, 838z Org. Syn. Coll. Vol. 5, 277References and Notes1.University of California, Los Angeles, California.2.Morton and Redman, Ind. Eng. Chem., 40, 1190 (1948).3.Stoll and Rouvé, Helv. Chim. Acta, 30, 1822 (1947).4.Prelog, Frenkiel, Kobelt, and Barman, Helv. Chim. Acta, 30, 1741 (1947).5.Stoll and Hulstkamp, Helv. Chim. Acta, 30, 1815 (1947).6.Hansley (to E. I. du Pont de Nemours and Company), U. S. pat. 2,228,268 [C. A., 35, 2534(1941)].7.Blomquist, Burge, and Sucsy, J. Am. Chem. Soc., 74, 3636 (1952).8.Prelog, Schenker, and Günthard, Helv. Chim. Acta, 35, 1598 (1952).AppendixChemical Abstracts Nomenclature (Collective Index Number);(Registry Number)Sebacoinmethyl or ethyl sebacateacetic acid (64-19-7)sodium carbonate (497-19-8)nitrogen (7727-37-9)sodium (13966-32-0)xylene (106-42-3)Pentane (109-66-0)magnesium sulfate (7487-88-9)Cyclodecanone, 2-hydroxy- (96-00-4)dimethyl sebacate (106-79-6)Copyright © 1921-2005, Organic Syntheses, Inc. All Rights Reserved。
氰转化为酰胺-060123
经典化学合成反应标准操作氰基转化为酯和酰胺目录1.前言 (2)2.氰基转化为酯 (2)3.氰基转化为酰胺 (2)3.1丙稀酰胺的合成 (2)3.2苯乙酰胺的合成 (3)6. 从氰基合成酰胺6.1氰基水解腈加水可以分解为伯酰胺。
由于伯酰胺会继续水解为羧酸,一般要控制水解的条件。
目前有许多方法报道,有时需要根据底物的特性选择酸性,碱性或中性的水解条件。
作为中性的条件,也有文献报道使用镍或钯催化剂的方法。
在酸性条件下与饱和碳相连的氰基,可以在酸中很方便的水解转化为酰胺,并在条件较为剧烈时,很容易进一步水解成酸。
但乙烯基或芳基腈的水解条件则要求剧烈得多,一般需要强酸条件,而且一般不会进一步水解。
在碱性条件下,利用过氧化氢氧化的方法可在室温下短时间内水解腈为伯酰胺,这是一个较为可靠的方法。
利用NaOH(aq.)-CH2Cl2相转移催化体系,DMSO-K2CO3体系[2]可以用于各种腈水解为伯酰胺。
6.1.1 盐酸水解腈为伯酰胺示例[3]HCl, H2OCN CONH2In a 3-l. three-necked round-bottomed flask equipped with glass joints are placed 200 g. (1.71 moles) of benzyl cyanide and 800 ml. of 35% hydrochloric acid. The flask is fitted with a reflux condenser, a thermometer, and an efficient mechanical stirrer. At a bath temperature of about 40° the mixture is stirred vigorously. Within a period of 20–40 minutes the benzyl cyanide goes into solution. During this time, the temperature of the reaction mixture rises about 10°above that of the bath. The homogeneous solution is kept in the bath with, or without, stirring for an additional 20–30 minutes. The warm water in the bath is replaced by tap water at about 15–20°, and the thermometer is replaced by a dropping funnel from which 800 ml. of cold distilled water is added with stirring. After the addition of about 100–150 ml., crystals begin to separate. When the total amount of water has been added, the mixture is cooled externally with ice water for about 30 minutes. The cooled mixture is filtered by suction. Crude phenylacetamide remains on the filter and is washed with two 100-ml. portions of water. The crystals are then dried at 50–80°. The yield of crude phenylacetamide is 190–200 g. (82–86%).6.1.2 浓硫酸水解不饱和腈为伯酰胺示例[4]CN CONH 21. H 2SO 42. NH 3To 106 g of 84 % sulfuric acid, was added 50 g of acrylonitrile. After stirring for 30 min at r.t., the resulting mixture was heated to 95 ℃, and stirred for 2 h. After cooling, the solid was collected by suction, and the filter cake was transferred into a beaker. To the ice-cooled solid, was added aq. ammonia with the speed that keep the temperature less than 50℃. The precipitated ammonium sulphate was filtered off, and the filtrate was cooled. The precipitate was collected by filtration, and the filter cake was washed by water, dried in vacuum to give the desired product.6.1.3 H 2O 2-K 2CO 3-DMSO 体系水解腈为伯酰胺示例[1] Cl CN30% H 2O 2, K 2CO 3DMSO, rt, 5 min ClONH 2To a stirred solution of 4-chlorobenzonitrile (1.37 g, 0.01 mol) in DMSO (3 ml), cooled in a ice bath, was added 30% H 2O 2 (1.2 ml) and K 2CO 3, the reaction was allowed to warm up to r.t. (strong exothermic effect was observed). After 5 min., distilled water (50 ml) was added, cooling applied, and the product was collected by filtration, yield 85%.6.1.4 NaOH(aq.)-CH 2Cl 2相转移催化体系水解腈为伯酰胺[2] CN (n -C 4H 9)N +HSO 4-30 % H O , CH Cl NH 2OTo a magnetically stirred dichloromethane solution (1.5 ml) of o -tolunitrile (0.5 g, 4.27 mmol) cooled in an ice ba th, are added 30% hydrogen peroxide (2.0 ml), tetrabutylammonium hydrogen sulfate (0.290 g, 0.85 mmol), and a 20% aqueous solution of sodium hydroxide (1.6 ml). Thereaction mixture is allowed to warm up to r.t. and maintained under stirring. After 1.6 h, dichloromethane is added, the organic layer is separated, washed with brine, and dried with sodium sulphate. The solvent is removed under reduced pressure to leave a white solid from which pu re o-toluamide is obtained by chromatography on silica gel. Yield 0.485 g (97%).6.2 Ritter反应碳正离子加成到腈基的氮原子上生成的腈盐加水分解得到相应的酰胺加水可以分解为酰胺。
Organic Syntheses, Coll. Vol. 4, p.218 (1963); Vol. 36, p.14 (1956).
Organic Syntheses, Coll. Vol. 4, p.218 (1963); Vol. 36, p.14 (1956).CYCLODECANONESubmitted by Arthur C. Cope, John W. Barthel, and Ronald Dean Smith 1.Checked by N. J. Leonard and J. C. Little. 1. ProcedureA 1-l. round-bottomed three-necked flask is fitted with a sealed stirrer (Note 1), a dropping funnel, and a reflux condenser, through which a thermometer extends nearly to the bottom of the flask. In the flask are thoroughly mixed 40.5 g. (0.62 g. atom) of zinc dust (Note 2) and 100 ml. of glacial acetic acid , and to this mixture is added 42.5 g. (0.25 mole) of sebacoin (p.840) (Note 3). The mixture is stirred rapidly, and 90 ml. of concentrated C.P. hydrochloric acid is added dropwise during a period of 5 to 10 minutes, or as fast as control of foaming and temperature permits. The temperature must be kept between 75 and 80° (Note 4), and cooling by a water bath may be necessary during the addition of the hydrochloric acid . Stirring is continued for 1.5 hours at 75–80°. Thirty minutes after the initial addition of hydrochloric acid , and again 30 minutes later, 90-ml. portions of concentrated hydrochloric acid are added to the mixture while the temperature is maintained at 75–80°. After the reaction is complete, the remaining zinc is separated from the cooled mixture by decantation (Note 5). The liquid phase is diluted with 700 ml. of saturated aqueous sodium chloride solution and extracted with four 250-ml. portions of ether , each of which is first used to wash the residual zinc (Note 6). The ether extracts are combined and washed with 250 ml. of saturated sodium chloride solution, three 250-ml. portions of 10% sodium carbonate solution (foaming!), and finally 250 ml. of saturated sodium chloride solution. The ethereal solution is dried over anhydrous magnesium sulfate (about 25 g. is needed). After the drying agent has been removed by filtration and the solvent by distillation, the residue is distilled at reduced pressure through an efficient column (Note 7). After a small fore-run consisting mostly of cyclodecane ,cyclodecanone is collected at 99–101°/8 mm. The yield is 29–30 g. (75–78%), n 25 1.4808–1.4810 (Note 8).2. Notes1. A metal stirrer must not be used. A simple glass stirrer with a ball-joint seal is satisfactory.2. Mallinckrodt technical grade may be used.2 If Mallinckrodt analytical reagent zinc dust is used, the reaction temperature must be maintained at 50–55° instead of 75–80°.3. Pure sebacoin gives a colorless product. A sebacoin-sebacil mixture must first be purified by recrystallization from pentane as described (p.841). The sebacil apparently is not reduced completely according to the accompanying directions and thus may contaminate the product (Note 7).4. The reaction temperature is important. At temperatures below 75° some sebacoin remains unreduced, while at temperatures above 80° considerable cyclodecane is formed. The submitters report that the reaction run at the reflux temperature gives cyclodecanone in 27% yield and cyclodecane in 32% yield.5. The product should be isolated and distilled as quickly as possible inasmuch as the unreacted sebacoin is readily oxidized to sebacil , which cannot be separated from the cyclodecanone by simple distillation.6. The residual zinc may be pyrophoric.7. For efficient separation of cyclodecanone from cyclodecane , a 60-cm. column of the simple Podbielniak type 3 may be used. Removal of sebacil cannot be accomplished readily by fractional distillation, since cyclodecanone and sebacil have virtually identical boiling points.8. Cyclodecanone regenerated from its semicarbazone, m.p. 203.5–205.5°, has n 25 1.4806.3. DiscussionD DThe procedure described is a modification of the directions of Prelog, Frenkiel, Kobelt, and Barman.4Cyclodecanone has been prepared by the dehydration of sebacoin followed by catalytic hydrogenation,5 by the pyrolysis of the thorium or yttrium salt of nonane-1,9-dicarboxylic acid,6 and by the ring enlargement of cyclononanone,7 as well as by the reduction of sebacoin.8This preparation is referenced from:z Org. Syn. Coll. Vol. 5, 277References and Notes1.Massachusetts Institute of Technology, Cambridge 39, Massachusetts.2.Brown and Borkowski, J. Am. Chem. Soc., 74, 1901 (1952).3.Cason and Rapoport, Laboratory Text in Organic Chemistry, 2nd ed., p. 294, Prentice-Hall,Englewood Cliffs, New Jersey, 1962.4.Prelog, Frenkiel, Kobelt, and Barman, Helv. Chim. Acta, 30, 1741 (1947).5.Stoll, Helv. Chim. Acta, 30, 1837 (1947).6.Ruzicka, Stoll, and Schinz, Helv. Chim. Acta, 9, 249 (1926); 11, 670 (1928).7.Kohler, Tishler, Potter, and Thompson, J. Am. Chem. Soc., 61, 1057 (1939).8.Blomquist, Burge, and Sucsy, J. Am. Chem. Soc., 74, 3636 (1952).AppendixChemical Abstracts Nomenclature (Collective Index Number);(Registry Number)Sebacoinsebacoin-sebacilthorium or yttrium salt of nonane-1,9-dicarboxylic acidhydrochloric acid (7647-01-0)acetic acid (64-19-7)ether (60-29-7)sodium chloride (7647-14-5)sodium carbonate (497-19-8)zinc (7440-66-6)Pentane (109-66-0)magnesium sulfate (7487-88-9)Cyclodecanone(1502-06-3)cyclodecane (293-96-9)cyclononanone (3350-30-9)Sebacil (96-01-5) Copyright © 1921-2005, Organic Syntheses, Inc. All Rights Reserved。
库尔提斯重排反应
库尔提斯重排反应[编辑](重定向自Curtius重排反应)库尔提斯重排反应(Curtius重排反应)是一个重排反应,首先由库尔提斯(Theodor Curtius)发现,反应中酰基叠氮重排生成异氰酸酯。
[1][2]关于此反应的综述参见:[3][4]。
产物可与一系列亲核试剂反应:与水作用水解得到胺;[5]与苯甲醇反应生成带有苄氧羰基保护基(Cbz)的胺类;[6]与叔丁醇作用生成带有叔丁氧羰基保护基(Boc)的胺类,用作有机合成中的重要中间体。
[7][8]羧酸1可通过与叠氮磷酸二苯酯2反应被转化为酰基叠氮3。
[9][10][11][12]目录[隐藏]∙ 1 反应机理∙ 2 延伸∙ 3 参考资料∙ 4 参见反应机理[编辑]反应中,酰基叠氮失去氮气生成酰基乃春(氮烯)2,然后烃基迅速迁移,生成产物异氰酸酯3:延伸[编辑]在Curtius重排反应的基础上,Darapasky递降反应(A. Darapsky, 1936)以α-氰基酯为原料,通过重排反应生成氨基酸。
[13]参考资料[编辑]1. ^ Curtius, T. Ber.1890, 23, 3023.2. ^ Curtius, T. J. Prakt. Chem.1894, 50, 275.3. ^ Smith, P. A. S. Org. React.1946, 3, 337-449. (Review)4. ^ Scriven, E. F.; Turnbull, K.; Chem. Rev.1988, 88, 297-368. Review5. ^ Kaiser, C.; Weinstock, J. Organic Syntheses, Coll. Vol. 6, p.910 (1988); Vol. 51,p.48 (1971).Article6. ^ Ende, D. J. a.; DeVries, K. M.; Clifford, P. J.; Brenek, S. J. Org. Proc. Res.Dev.1998, 2, 382-392.7. ^ Lebel, H.; Leogane, O.; Org. Lett.2005, 7(19),4107-4110. doi:10.1021/ol051428b8. ^ Shioiri, T.; Yamada, S. Organic Syntheses, Coll. Vol. 7, p.206 (1990); Vol. 62,p.187 (1984).Article9. ^ Shioiri, T.; Ninomiya, K.; Yamada, S. J. Am. Chem. Soc.1972, 94,6203-6205.doi:10.1021/ja00772a05210. ^ Ninomiya, K.; Shioiri, T.; Yamada, S. Tetrahedron1974, 30, 2151-2157.11. ^ Wolff, O.; Waldvogel, S. R. Synthesis2004, 1303-1305.12. ^ Jessup, P. J.; Petty, C. B.; Roos, J.; Overman, L. E. Organic Syntheses, Coll.Vol. 6, p.95 (1988); Vol. 59, p.1 (1979). Article13. ^/reactions/RXN051.htm(重定向自贝克曼重排)贝克曼重排反应(Beckmann rearrangement)是一个由酸催化的重排反应,反应物肟在酸的催化作用下重排为酰胺。
酯化以及酰胺化
HOOCCOOHOHOH2TsOH, toluene2CH2Ph2CH2PhA 300-mL, one-necked, round-bottomed flask was equipped with a magnetic stirrer, Dean-Stark trap, and a reflux condenser. The flask was charged with 3.0 g (20 mmol) of L-(+)-tartaric acid, 6.5 g (60 mmol) of benzyl alcohol, 47.5 mg (0.25 mmol) of p-toluenesulfonic acid monohydrate and 40 mL of toluene. The mixture was heated under reflux in an oil bath (about 130℃) for 13 h. During this period the theoretical amount of water (0.62 mL) was collected. The mixture was allowed to cool to ambient temperature, diluted with ether, and poured into 50 mL of aqueous, saturated sodium bicarbonate. The organic phase was separated and the aqueous phase was extracted twice with 20 mL of ether. The combined organic phases were dried over sodium sulfate. The solvent was removed with a rotary evaporator, and the resulting crude product was triturated with hexane-ether (20:1, 210 mL) to give white crystals of (−)-dibenzyl tartrate. The precipitate was collected by filtration and washed with hexane-ether (20:1). The filtrate was further concentrated to give a second crop. The total yield was 6.2 g (94%), mp 49–50℃[5]2.5 酰氯和醇、酚的酯化反应示例:酰氯是强酰化剂和醇、酚作用生成酯的反应很迅速,此方法适用于由空间障碍的酯化。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
Organic Syntheses, Coll. Vol. 3, p.207 (1955); Vol. 21, p.18 (1941). CHOLESTENONE
Submitted by R. V. Oppenauer Checked by J. Cason and L. F. Fieser.
1. Procedure A carefully dried 5-l. round-bottomed flask equipped with a reflux condenser carrying a calcium chloride tube is charged with 100 g. of cholesterol (Note 1), 750 ml. of acetone (Note 2), and 1 l. of benzene (Note 3). A boiling tube is introduced to prevent bumping (Note 4), and the mixture is heated to boiling in an oil bath which is maintained at 75–85° during the reaction. A solution of 80 g. of aluminum tert-butoxide (p. 48) in 500 ml. of dry benzene is added in one portion to the boiling solution. The mixture turns cloudy and in 10–15 minutes develops a yellow color. Gentle boiling is continued at a bath temperature of 75–85° for a total of 8 hours. The mixture is then cooled, treated with 200 ml. of water and then 500 ml. of 10% sulfuric acid, shaken vigorously, and transferred to a 5-l. separatory funnel. The mixture is diluted with 1.5 l. of water and shaken for several minutes, after which the yellow aqueous layer is drawn off into a second separatory funnel and shaken out with a small amount of benzene (Note 5). The combined benzene extracts are washed thoroughly with water and dried by filtration through a layer of sodium sulfate; the solvent is evaporated, the last traces being removed by heating the residue at 60° at the water pump vacuum. The oily yellow residue solidifies when it is cooled in an ice-salt bath and scratched. For crystallization the material is dissolved in a mixture of 70 ml. of acetone and 100 ml. of methanol; the solution is allowed to cool very slowly and is seeded, for otherwise the product tends to separate as an oil. After the bulk of the material has crystallized, the mixture is allowed to stand for 1 day at 0°; the product is then collected, washed with 100 ml. of ice-cold methanol, and dried in vacuum at room temperature. The yield of almost colorless cholestenone, m.p. 77–79°, is 70–81 g. (70–81%). Recrystallization by the same method gives material melting at 78.5–80.5° with 90% recovery (Note 6) and (Note 7).
2. Notes 1. The material should be dried to constant weight at 80–100° in vacuum. Commercial cholesterol, m.p. 146–148.5°, gives satisfactory results; the yield is raised 5–10% by using cholesterol that has been purified by regeneration from the dibromide. 2. The acetone is distilled once from permanganate and twice from freshly fused potassium hydroxide. 3. The benzene is distilled over sodium. 4. If boiling stones are employed, fresh ones must be added at intervals; the boiling tube promotes smooth boiling at the bath temperature specified. 5. The troublesome emulsions sometimes encountered are easily broken by filtration from a trace of suspended solid. 6. For recovery of material in the mother liquors, the solutions are subjected to steam distillation to remove solvents and condensation products from the acetone. The process is continued until the distillate is odorless (1–2 hours), and the residual oil is dried by heating it under reduced pressure on the steam bath. From a solution in acetone-methanol (20 ml. and 50 ml., respectively) there is obtained after some manipulation (slow cooling, scratching) 3.3 g. of crude cholestenone, m.p. 72–80° (cloudy melt). 7. The yield is reduced by about 5% by halving the amounts of acetone and benzene specified and using only 50 g. of aluminum tert-butoxide. 3. Discussion Cholestenone has been prepared by oxidation of cholesterol dibromide with chromic acid1,2 or potassium permanganate3 and subsequent debromination; by dehydrogenation of cholesterol over copper oxide;4,5 and by the method described above.6,7
This preparation is referenced from:
zOrg. Syn. Coll. Vol. 4, 192 zOrg. Syn. Coll. Vol. 4, 195
References and Notes 1.Windaus, Ber., 39, 518 (1906). 2.Bretschneider and Ajtai, Monatsh., 74, 57 (1943). 3.Schoenheimer, J. Biol. Chem., 110, 461 (1935). 4.Diels and Abderhalden, Ber., 37, 3099 (1904). 5.Diels, Gädke, and Körding, Ann., 459, 21 (1927). 6.Oppenauer, Rec. trav. chim., 56, 137 (1937). 7.Brit. pat. 487,360 [C. A., 33, 323 (1939)].
Appendix Chemical Abstracts Nomenclature (Collective Index Number); (Registry Number)
sulfuric acid (7664-93-9) Benzene (71-43-2) methanol (67-56-1) potassium permanganate (7722-64-7) sodium sulfate (7757-82-6) acetone (67-64-1) potassium hydroxide (1310-58-3) sodium (13966-32-0) chromic acid (7738-94-5) copper oxide (1317-38-0) Cholesterol (57-88-5) Cholestenone