聚乙烯的紫外交联 - 江南大学杂志社
0聚丙烯酰胺交联微球的制备及其粒径影响因素

性能,PAMCMS在水中能长时间保持溶胀。
[关键词]丙烯酰胺;聚丙烯酰胺交联微球;反相悬浮聚合;溶胀性能;油田调剖堵水剂
[文章编号]1000—8144(2008)10—1059一05
[中图分类号]TQ 322.4
[文献标识码]A
Preparation and Influential Parameters of Crosslinked Polyacrylamide Microspheres
近的强吸收峰基本消失,表明AM具有良好的聚合
活性。
万方数据
图1 AM(a)和PAMCMS(b)的FTIR谱图
PAMCMS(b) Fig.1 FHR spectra of AM(a)and
2.2 影响PAMCMS粒径的因素 2.2.1 搅拌转速的影响
固定各组分用量不变(见表1),考察搅拌转速
对PAMCMS平均粒径的影响,实验结果见图2。由 图2可看出,随搅拌转速的提高,PAMCMS的粒径
change.PAMCMS can be used as water
plugging materials in oil exploitation.
[Keywords] acrylamide;polyacrylamide crosslinking microsphere;inverse suspension polymeriza— tion;swelling property;profile controlling—water plugging agent for衄field
studied.Under the conditions:reaction temperature 50—65 oC,reaction time 4 h and N2 atmosphere,
聚乙烯的热降解动力学研究

聚乙烯的热降解动力学研究黄钰香;庞承焕;吴博;张云【摘要】采用三种不同的动力学分析方法,即Freeman方法、Flynn - Wall - Ozawa以及Kissinger方法对不同类型聚乙烯的热分解动力学进行了探讨.结果表明,Flynn - Wall - Ozawa法Ⅰ、Friedman法的测试结果与三者聚乙烯的结构特征较吻合,不同聚乙烯降解活化能的大小顺序为HDPE> LLDPE> LDPE.%Three analysis methods including the Freeman method,Flynn-Wall-Ozawa method and Kissinger method were used to investigate the thermal degradation kinetics of High-density polyethylene ( HDPE) and Low-density polyethylene (LDPE) and low-density polyethylene ( LLDPE ). The results obtained by the Flynn-Wall-Ozawa I method and Friedman method are close to the structural characteristics of polyethylene and it has been found that the activation energies of HDPE are higher than the value of LLDPE.【期刊名称】《合成材料老化与应用》【年(卷),期】2012(041)004【总页数】7页(P9-15)【关键词】热重;热降解;热降解动力学;活化能;聚乙烯【作者】黄钰香;庞承焕;吴博;张云【作者单位】金发科技股份有限公司产品研发中心,塑料改性与加工国家工程实验室,广东广州510500;金发科技股份有限公司产品研发中心,塑料改性与加工国家工程实验室,广东广州510500;金发科技股份有限公司产品研发中心,塑料改性与加工国家工程实验室,广东广州510500;广州合成材料研究院有限公司,广东广州510665【正文语种】中文【中图分类】TQ325.1+2聚合物的热分解特性一直是人们研究的焦点,这方面的文献报道很多[1-4].由于聚合物结构的复杂性,聚合物的热分解比通常的小分子无机化合物要复杂得多。
二步法硅烷交联聚乙烯配方试验总结

杭州通达高分子材料有限公司试验记录二步法硅烷交联聚乙烯配方试验总结2009 年 11 月本试验的目的是采用价格较低的LLDPE (吉林石化, 7042)代替LDPE 作为二步法Si-XLPE 的主要原材料进行配方设计与优化,以期能减少配方成本。
主要原材料树脂如表 1 所示:表格 1 二步法硅烷交联聚乙烯试验用树脂列表树脂类型熔融指数 ,190℃价格 /元( g/10min )(11 月 18 日)吉林石化 7042 LLDPE 1.74 10,900福建联合 8320 LLDPE 20.3 11,400 沙特 6101 LLDPE 20中海壳牌 2426H LDPE 1.83 12,400 伊朗 0190 LDPE 1.781.小样试验工艺及参数的确定为了保证试验方法的准确性,排除因试验条件的不一致而对试验结果产生不利影响,对小试工艺进行了调整,采用了不同的工艺及设备进行试验最终确定一种合理的试验方法。
1.1 试验一:利用与哈普杂质检测仪连接的小型单螺杆挤出机放片(11月 4日~7日)按表 2 配方将混合树脂与硅烷油装入铝塑袋,热封后混匀放置一夜。
按表 3 中工艺设定在哈普杂质检测仪的挤出机上放片, A 料与 B 料按 95:5 投放。
表格 2 A 料配方表 (Ⅰ)份编S1101 S1102 S1103 S1104 数号配方7042 90 85 85 80 6101 10 15 10 10 2426H —— 5 10 硅烷油 1.3 1.35 1.35 1.35表格 3 工艺设定 (Ⅰ)1 区2 区3 区4 区口模转速130℃145℃155℃160℃165℃400rpm结果与分析:物料从口模出来明显的流动性太大,经过辊压后料片非常薄(约0.2mm),并且随着混合 LLDPE 中高熔融指数(6101)比例的增加,物料的流动性更大,温度不到165℃已经不易加工;随着2426H 含量的增加,料片的表面光洁度得到改善。
pH响应性聚乙二醇-co-聚丙烯酸交联微球的合成

第5卷第6期2006年12月 江南大学学报(自然科学版)Journal of Southern Yangtze U niversity(N atural Science Edition) Vol.5 No.6Dec. 2006 文章编号:1671-7147(2006)06-0730-04 收稿日期:2005-11-15; 修订日期:2006-01-10. 基金项目:国家自然科学基金资助项目(50443012). 作者简介:倪忠斌(1969-),江苏南通人,工程师,工学硕士.主要从事功能材料的制备研究.Email :mqchen @p H 响应性聚乙二醇Οco Ο聚丙烯酸交联微球的合成倪忠斌, 张 坤, 朱雪艳, 陈明清, 刘晓亚, 杨 成(江南大学化学与材料工程学院,江苏无锡214122)摘 要:在N ,N ′Ο亚甲基二丙烯酰胺(Bis )存在的条件下,以偶氮二异丁腈(A IBN )为引发剂,使苯乙烯单封端聚乙二醇(St ΟPEG )大分子单体与丙烯酸(AAc )在3Ο戊酮溶液中进行分散共聚反应,得到了聚乙二醇Οco Ο聚丙烯酸(PEG Οco ΟPAAc )交联微球.动态激光光散射测试结果表明,所得的交联微球具有良好的单分散性;同时发现,在共聚反应中St ΟPEG 大分子单体、AAc 和Bis 溶液浓度对交联微球粒径有明显的影响;交联微球的R h 在p H 为4.5附近突然增加,并在p H >7.0后保持不变,说明该PEG Οco ΟPAAc 交联微球具有明显的p H 响应性.关键词:丙烯酸;聚乙二醇大分子单体;分散共聚;交联微球;p H 响应性中图分类号:O 641文献标识码:APreparation of Crosslinking Poly(Ethylene G lycol)Οco ΟPoly(Acrylic Acid)Microspheres with p H ΟR esponsive PropertiesN I Zhong Οbin , ZHAN G Kun , ZHU Xue Οyan , CH EN Ming ΟqingL IU Xiao Οya , YAN G Cheng(School of Chemical and Materials Engineering ,Southern Yangtze University ,Wuxi 214122,China )Abstract :The p H Οresponsive cro sslinking poly (et hylene glycol )Οco Οpoly (acrylic acid )(PEG Οco ΟPAAc )micro sp heres were p repared by dispersion copolymerization of St ΟPEG macromonomer wit h acrylic acid in 3Οpentanone in t he present of N ,N’Οmet hylenebisacrylamide (Bis )as crosslinker and 2,2’ΟAzobis (isobutyronitrile )(A IBN )as initiator.The dynamic laser light scattering (DL S )measurement showed t hat t he cro sslinking microsp heres have narrow size dist ribution.The effect s of reaction parameters such as AAc concent ration ,PEG macromonomer concent ration and Bis concent ration on t he R h of obtained cro sslinking microsp heres were investigated.It was also found t hat t he PEG Οco ΟPAAc crosslinking micro sp heres behave excellent p H Οresponsive and exhibit a rapid increase in R h when t he p H value varies f rom about 4.5to 7.0.K ey w ords :acrylic acid ;St ΟPEG macromonomer ;dispersion copolymerization ;crosslinking micro sp here ;p H Οresponsive 聚合物微球具有比表面积大、表面活性高和易于分离回收等特点,广泛应用于生物医学、高效催化和环境保护等高新技术领域[1Ο3].其中,智能型微球以其受到环境刺激时快速发生突变,呈现相转变行为的特性,引起了各国化学和生物医学工作者的极大兴趣[4].目前,对智能型微球的研究主要集中在p H敏感性的聚甲基丙烯酸类及温敏性的聚(NΟ异丙基丙烯酰胺)类交联微球或微凝胶的制备及应用[4Ο6].其制备大多采用沉淀聚合或反相乳液聚合,由于聚合过程中加入的乳化剂或分散剂在体系中的残留将对其在药物释放等领域中的应用产生影响.G oh 等[7]曾以乙二醇二甲基丙烯酸酯为交联剂,采用分散共聚的方法制备了交联度极高(交联剂与甲基丙烯酸单体物质的量比大于5∶2)的PEGΟcoΟPMAA交联微球,由于较高的交联度使微球难以被溶胀,故微球失去了对环境刺激的响应能力.文中选用丙烯酸(AAc)单体,以N,N′Ο亚甲基二丙烯酰胺(Bis)为交联剂,在3Ο戊酮(L P)溶液中与苯乙烯单封端聚乙二醇(StΟPEG)大分子进行自由基分散共聚反应,制备低交联度的聚乙二醇ΟcoΟ聚丙烯酸(PEGΟcoΟPAAc)交联微球.并考察反应体系中StΟPEG大分子单体、单体AAc、交联剂Bis和引发剂偶氮二异丁腈(A IBN)浓度对交联微球粒径的影响,进而对所得PEGΟcoΟPAAc交联微球的p H 响应性进行研究.1 材料与方法1.1 试剂与设备1.1.1 试剂 AAc,分析纯,上海化学试剂公司生产,减压蒸馏后使用;StΟPEG(M w=5010(M w/M n= 1.05),末端官能团(C=C)含量为97.5%),以PEG (MeΟOΟPEGΟO H)低聚物(由日本油脂公司提供)为原料,根据文献[8]的方法合成得到;AI BN,日本和光公司提供,经乙醇重结晶提纯后使用;Bis,日本和光公司提供;L P,上海试剂一厂生产.1.1.2 设备 Agilent1100型凝胶色谱仪(GPC),美国Agilent公司制造;Bruker DMX500,500 M Hz型核磁共振仪(NMR),英国Bruker公司制造;ALVΟ5000e动态激光光散射仪(DL S),德国ALV公司制造.1.2 聚合物微球的合成将1.4mmol AAc、0.028mmol StΟPEG大分子单体及与AAc的物质的量比为1%的AI BN和Bis 置于样品瓶中,加入5mL3Ο戊酮溶液作溶剂,使StΟPEG大分子单体和AI BN充分溶解后,将溶液移入聚合反应管中,并用高纯N2置换5min后封管.将反应管置于60℃恒温振动水浴(150r/min)中反应24h,得交联聚合物微球分散液.所得样品以丙酮溶液为洗涤剂,经数次离心分离除去未反应单体,最后分散得PEGΟcoΟPAAc微球分散液.PEGΟcoΟPAAc交联微球的合成反应式见图1.图1 PEGΟcoΟPAAc交联微球的合成Fig.1 Synthesis of PEGΟcoΟPAAc crosslinkingmicrospheres1.3 测试方法大分子单体的相对分子质量以及相对分子质量分布由GPC(Agilent1100PL gel分离柱)测定,以聚乙二醇为标样,其流动相为DM F,体积流量为0.6mL/min,温度为35℃.大分子单体的末端官能团(C=C)含量由1HΟNMR(Bruker DMX500,500 M Hz)测定得出.微球的流体力学半径(R h)及其分布由DL S测试得出,测定温度为(25±0.1)℃,相关器为ALVΟ5000e,激光波长632.8nm,散射角90°,R h及其分布均由CON TIN模拟获得.2 结果与讨论2.1 PEGΟcoΟPAAc微球的合成根据大分子单体参与的分散共聚反应机理:反应前,AAc单体、StΟPEG大分子单体、交联剂Bis 和引发剂A IBN完全溶解在反应介质(3Ο戊酮溶液)中;随着引发剂的分解引发单体聚合得到的PAAc 链段因不溶于反应介质并在聚合物链段间形成强烈的氢键相互作用而发生聚集[7],交联剂的存在则使聚集体内部发生交联,同时共聚物中的PEG链段则因其在反应介质中具有良好的溶解性而对PAAc链段的聚集体(核)起到稳定作用,形成稳定微球.图2为反应得到的PEGΟcoΟPAAc交联微球的动态激光光散射测试结果.可以看出,在有交联剂存在的条件下得到的微球具有较窄的粒径分布,且其平均粒径也较无交联剂体系中得到的微球小.由此可见,在本反应体系中,添加适量的交联剂Bis137 第6期倪忠斌等:p H响应性聚乙二醇ΟcoΟ聚丙烯酸交联微球的合成溶液一方面可改善颗粒的单分散性,另一方面可通过改变Bis 溶液的用量来控制微球大小.图2 PEG Οco ΟPAAc 微球的R h 分布Fig.2 R h distribution of crosslinking PEG Οco ΟPAAcmicrospheres2.2 大分子单体用量对交联微球粒径的影响固定AAc 浓度并保持Bis 与单体的物质的量比为0.01,改变St ΟPEG 大分子单体浓度,所得交联微球的流体力学半径(R h )与St ΟPEG 大分子单体浓度的关系如图3所示.可以看出,随着St ΟPEG 浓度的增加,微球的R h 逐渐减小,这是由于PEG 链段在分散共聚中起着稳定剂的作用.随着大分子单体浓度的增加,共聚物中亲溶剂性PEG 链段含量也相应增加,在成核初期能稳定更多的核,使体系中核的数量增加;同时亲溶剂性PEG 链段可以保护微球稳定地分散于介质中而不发生凝聚,所以最终可得到R h 较小的微球.另一方面,根据吴奇等[8]的单个大分子链所能稳定的微球表面积一定的结论,增加大分子单体的加入量,也使体系倾向于形成粒径较小的微球.图3 交联微球的R h 与St ΟPEG 浓度的双对数关系Fig.3 Double Οlogarithmic plots of the radius ofcrosslinking microspheres vs [St ΟPEG ]2.3 AAc 单体浓度对交联微球粒径的影响由图3还可看出,大分子单体St ΟPEG 、交联剂Bis 和引发剂A IBN 的浓度一定时,所得交联微球的R h 随体系中丙烯酸浓度的增加而增加.一方面,丙烯酸浓度的增加会改变反应介质的极性,减弱PAAc 链段间氢键的相互作用,使反应初期产生的聚合物临界链长增大,导致体系在成核过程中形成稳定微核数量减少,从而得到粒径较大的微球.另一方面,大分子单体量一定时,其所能稳定的总表面积也为一常数[8];AAc 单体加入量增加,则会使体系倾向于形成比表面积较小、粒径较大的微球.2.4 引发剂浓度对交联微球粒径的影响固定St ΟPEG 大分子单体浓度(0.01mol/L )、AAc 浓度(0.69mol/L ),交联剂Bis 与单体的物质的量比为0.01不变,引发剂A IBN 起始浓度对微球流体力学半径的影响如图4所示.引发剂起始浓度在0.003~0.028mol/L 的范围内,交联微球的粒径基本上不随A IBN 初始浓度的变化而变化.表明在该体系中,改变引发剂初始浓度并不能有效地改变交联微球粒径,这与一般的分散共聚体系有明显的差异[9].图4 交联微球的R h 与AIBN 浓度的双对数关系Fig.4 Double Οlogarithmic plots of the radius ofcrosslinking microspheres vs [AIBN ]2.5 交联剂浓度对交联微球粒径的影响固定St ΟPEG 大分子单体浓度(0.0056mol/L )、AAc 浓度(0.28mol/L )和A IBN 与单体的物质的量比为0.01不变,交联剂Bis 初始浓度对微球流体力学半径的影响如图5所示.可以看出,交联剂Bis 初始浓度由2.8mmol/L 增加到7mmol/L 时,微球的R h 也相应地从420nm 减小到200nm ,即交联微球的粒径随交联剂浓度增加而减小.主要原因是增加交联剂浓度可能会增加成核初期核间交联的几率,增加了核的紧密程度,也增加了反应介质中的单体扩散进入核内反应的阻力,使得微球的粒径减小.此外,PAAc 链段间形成的较强氢键也将增大PAAc 链段间的相互作用力,从而增加单体扩散进入微核的阻力,使得微球的粒径减小.2.6 PEG Οco ΟPAAc 交联微球对pH 的响应性图6为PEG Οco ΟPAAc 交联微球的R h 与分散介质p H 值的关系.可以看出,随着分散介质p H 值的增加,PEG Οco ΟPAAc 交联微球的R h 逐渐增大,且在p H 值为4.5左右时发生变化,该p H 值同聚丙237 江南大学学报(自然科学版) 第5卷 图5 交联微球的R h 与Bis 浓度的双对数关系Fig.5 Double Οlogarithmic plot of the radius ofcrosslinking microspheres vs [Bis]烯酸的p K a 值(4.75)基本一致.由DL S 所测数据分析计算,交联微球的R h 从p H =2时的420nm 增大到p H =4时的580nm ,继而迅速增加,到p H =6.86时可达1200nm ,微球体积增大27倍.这是因为,在体系p H 值高于聚丙烯酸的p K a 值时,聚丙烯酸发生电离,生成高分子的多羧酸根离子和氢离子(抗衡离子),呈离解平衡状态,共聚物整体表现出聚电解质的特性.共聚物中的聚丙烯酸链段的离解平衡态受到溶液p H 值的影响,即颗粒具有p H 响应性.当p H 值较低(抗衡离子浓度较大)时,氢离子在聚阴离子链的外部与内部进行扩散,使部分阴离子静电场得到平衡,以致其排斥力作用减弱,共聚物链段发生蜷曲,尺寸减小;当p H 值较高时,离解度增加,聚合物链段周围的抗衡离子浓度相对较小,聚合物链段上的阴离子与邻近的阴离子相互排斥力作用增加,使得聚合物链段在水溶液中呈伸展状态,因而颗粒尺寸增加.图6 交联微球的R h 与pH 值的关系Fig.6 R h of crosslinking microsphere vs pH3 结 论文中选用AAc 单体,以Bis 为交联剂、A IBN 为引发剂,与St ΟPEG 大分子单体进行自由基分散共聚反应,制备了低交联密度的PEG Οco ΟPAAc 交联微球.改变反应体系中St ΟPEG 大分子单体、单体AAc 和交联剂Bis 的浓度,可达到控制交联微球粒径的目的.试验还发现,当体系的p H 值从4增加到6.86时,交联微球的R h 可从580nm 增加到1200nm ,显示出良好的p H 响应性.该高分子交联微球具有完全的亲水性,同时具有p H 响应性和生理相容性,因而将会在生物医学研究领域中有广阔的应用前景.参考文献:[1]Sakuma S ,Suzuki N ,Kikuchi H ,et al.Oral peptide delivery using nanoparticles composed of novel graft copolymershaving hydrophobic backbone and hydrophilic branches[J ].Int J Pharm ,1997,149(1):93Ο106.[2]赖家平,曹现峰,何锡文.水溶液中制备分子印迹聚合物微球及其分子识别特性研究[J ].化学学报,2002,60:322Ο325.[3]CH EN Chun Οwei ,CH EN Ming Οqing ,Serizawa T ,et al.In situ synthesis and the catalytic properties of platinum colloidson polystyrene microspheres with surface Οgrafted poly (N Οisopropylacrylamide )[J ].Chem Commun ,1998,1,831Ο832.[4]朱雪燕,陈明清,刘晓亚,等.温敏性凝胶的合成与药物缓释模拟[J ].江南大学学报:自然科学版,2002,1(2):160Ο163.[5]陈兆伟,陈明清,刘晓亚,等.p H 快速响应PN IPAAm Οco ΟPAAc 水凝胶的制备及溶胀动力学[J ].应用化学,2004,21(9):884Ο889.[6]Eichenbaum G M ,Kiser P F ,Shah D ,et al.The dependence on functional group composition [J ].Macromolecules ,1999,32:8996Ο9006.[7]G oh E C C ,Stover H D H.A morphology study[J ].Macromolecules ,2002,35:9983Ο9989.[8]WU C ,Akashi M ,CH EN Ming Οqing.A simple structural model for the polymer microsphere stabilized by the PEOmacromonomers grafted on its surface[J ].Macromolecules ,1997,30:2187Ο2189.[9]CH EN Ming Οqing ,Serizawa T ,K ishida A ,et al.Particle size control of PEG coated polystyrene nanoparticles bymacromonomer method[J ].J Polym Sci :Part A :Polym Chem ,1999,37:2155Ο2166.(责任编辑:邢宝妹)337 第6期倪忠斌等:p H 响应性聚乙二醇Οco Ο聚丙烯酸交联微球的合成。
【江苏省自然科学基金】_交联结构_期刊发文热词逐年推荐_20140816
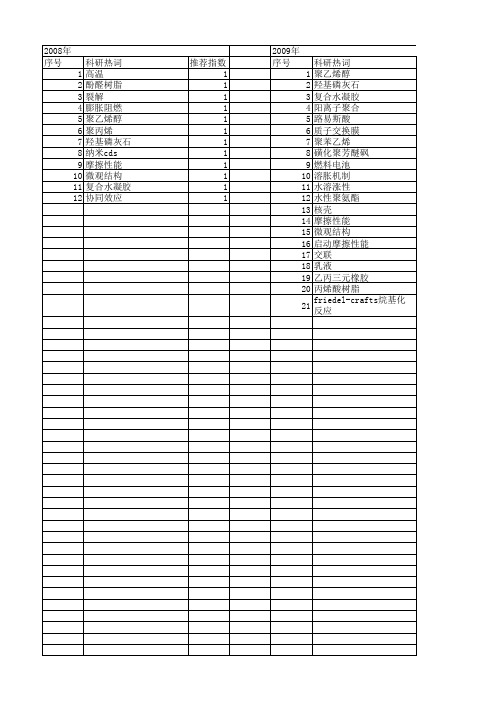
科研热词 高温 酚醛树脂 裂解 膨胀阻燃 聚乙烯醇 聚丙烯 羟基磷灰石 纳米cds 摩擦性能 微观结构 复合水凝胶 协同效应
推荐指数 1 1 1 1 1 1 1 1 1 1 1 1
2009年 序号 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21
科研热词 聚乙烯醇 羟基磷灰石 复合水凝胶 阳离子聚合 路易斯酸 质子交换膜 聚苯乙烯 磺化聚芳醚砜 燃料电池 溶胀机制 水溶涨性 丙烯酸树脂 friedel-crafts烷基化反应
推荐指数 2 2 2 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1
2012年 序号 1 2 3 4
科研热词 蓝藻废水 复合絮凝剂 psafc cts
推荐指数 1 1 1 1
2013年 序号 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19
科研热词 魔角变换-核磁共振 高分子化学 质子交换膜 聚丙烯酰胺 紫外透过性能 离子液体 磺化聚芳醚砜 硅磷酸盐 热性能 歧化松香 材料 无模板合成 弯曲性能 含氮碳材料 半互穿聚合物网络结构 前驱体 共聚材料 光学玻璃 乙烯基酯树脂
2010年 序号
科研热词 1 葡萄糖氧化酶 2 葡萄糖传感器 3 纳米氧化铋
推荐指数 1 1 1
2011年 序号 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29
2011年 科研热词 壳聚糖 高压脉冲电场 链霉菌 超支化 谷氨酰胺转胺酶 裂纹扩展 蛋白质表达 蛋白质结构 蛋清蛋白 药物缓释 聚磷酸钙 聚氨酯 结构 水分散液 氧化海藻酸钠 拉伸强度 微球 孔隙率 孔参数 多孔生物陶瓷 复合体系 吸附性 功能性质 力学性能 剪切强度 分子改造 凹凸棒黏土 乳化交联 pva水凝胶 推荐指数 2 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1
高分子材料

第一章1塑料和橡胶工业的发展历程——标志性的事件;(1)19世纪中叶第一种工业化的塑料----赛璐珞”(Celluloid)的塑料(1869)(最早被应用的塑料)(2)雷奥.比克兰德合成酚醛树脂(PF),第一个工业化生产的合成树脂(第一种人工合成树脂)(3)1920年,Staudinger首先提出了高分子的概念(4)Zieglar-Natta催化剂合成出了低压高密度聚乙烯(HDPE, 1953~1955)和聚丙烯(PP)(HDPE和PP的合成方法是谁发明的)(5)①1823年,苏格兰化学家马金托什,像印第安人一样把白色浓稠的橡胶液体涂抹在布上,制成防雨布,并缝制了“马金托什”防水斗蓬,这是世界上最早的雨衣,也是橡胶工业的起点②1826年,英国人汉考克发明了双辊开炼机,用此设备可以将各种助剂混入橡胶中;1839年,美国化学家固特异尔偶然中发明了橡胶的硫化,解决了橡胶遇热变软发粘的缺点,制造出了世界第一双橡胶防水鞋,这两项发明使橡胶的应用得到了突破性的进展,奠定了现代橡胶加工业的基础(是什么发现导致了近现代意义橡胶工业的诞生?)2高分子科学的进步对高分子材料工业发展的推动作用;通过硫化改性,有力地推动了橡胶工业的发展,因为硫化胶的性能比生胶优异得多,从而开辟了橡胶制品广泛应用的前景。
同时,橡胶的加工方法也在逐渐完善,形成了塑炼、混炼、压延、成型这一完整的加工过程,使得橡胶工业蓬勃兴起,发展突飞猛进。
合成高分子的诞生和发展是从酚醛树脂开始的,人类历史上第一个完全靠化学合成方法生产出来的合成树脂出现后,合成并工业化生产的高分子材料种类迅速发展。
3橡胶和塑料的基本概念橡胶:提取橡胶树、橡胶草等植物的胶乳,加工后制成的具有弹性、绝缘性、不透水和空气的一类线型柔性高分子材料。
塑料:利用单体原料以合成或缩合反应聚合而成高分子材料,由合成树脂及填料、增塑剂、稳定剂、润滑剂、色料等添加剂组成的。
4树脂与塑料、通用树脂与塑料;树脂:是指受热后有软化或熔融范围,软化时在外力作用下有流动倾向,常温下是固态、半固态,有时也可以是液态的有机聚合物。
透明质酸_胶原蛋白_聚乙二醇复合水凝胶的研究_袁静
文章编号:1001-9731(2015)15-15052-06透明质酸/胶原蛋白/聚乙二醇复合水凝胶的研究*袁静,邱立朋,陈敬华(江南大学药学院,江苏无锡214122)摘要:针对透明质酸、胶原蛋白水凝胶力学性能差的问题,采用两步交联法将透明质酸、胶原蛋白与聚乙二醇双丙烯酸酯制备成一种兼具生物相容性和高强度的复合水凝胶。
研究制备了具有相同胶原蛋白和聚乙二醇双丙烯酸酯质量分数,不同透明质酸质量分数的水凝胶,从微观结构和力学强度调控了复合水凝胶的理化性能,并用L-赖氨酸对复合凝胶进行改性。
SEM 结果表明,三元复合水凝胶具有蜂窝状的内部结构,孔径范围在80 180μm,赖氨酸浓度达40mmol/L时,水凝胶的压缩模量达414kPa,是改性前模量的19倍。
细胞毒性和体内植入实验表明该水凝胶浸提环境对细胞生长无明显抑制作用,产生较低的免疫排斥反应,有望用于软骨等组织的修复。
关键词:透明质酸;胶原蛋白;聚乙二醇双丙烯酸酯;水凝胶中图分类号:R318文献标识码:A DOI:10.3969/j.issn.1001-9731.2015.15.0101引言水凝胶具有三维网络的结构并且含水量高,这种结构组成与细胞生存的外环境十分相似。
因此,利用水凝胶来仿真胞外基质,为细胞在体外提供生长环境一直是组织工程研究的重点领域之一[1-3]。
目前制备医用水凝胶的主要天然高分子材料有胶原蛋白[4]、纤维蛋白[5]、明胶[6]、透明质酸[7]、硫酸软骨素[8]、壳聚糖[9]等。
这些天然大分子虽然具有很好的生物相容性,但与合成高分子材料相比,稳定性和机械性能差。
通常天然高分子水凝胶模量在几十kPa到几百kPa之间,而软骨组织的模量在0.1 1.0MPa[10],因此提高天然高分子水凝胶的力学性能在软骨和骨组织工程领域具有重要的意义。
Gong等通过两步法交联得到的2-丙烯酰胺-2-甲基丙磺酸(PAMPS)与丙烯酰胺(AM)双网络水凝胶具有优良的机械性能[11]。
20764_ftp
Preparation of Polymer/Silica Composite Nanoparticles Bearing Carboxyl Groups on the Surface viaEmulsifier-Free Emulsion CopolymerizationZHONG ZENG,JIAN YU,ZHAO-XIA GUOInstitute of Polymer Science and Engineering,Department of Chemical Engineering,School of Materials Science and Engineering,Tsinghua University,Beijing100084,ChinaReceived3November2004;accepted30January2005DOI:10.1002/pola.20764Published online in Wiley InterScience().ABSTRACT:Polymer/silica organic/inorganic composite nanoparticles bearing carboxylgroups on the surface were prepared via the emulsifier-free emulsion copolymeriza-tion of methyl methacrylate and sodium methacrylate(NaMA).Carboxyl groups weregenerated by the addition of hydrochloric acid at the end of the copolymerization.Two methods of NaMA addition were studied:batch and two-stage procedures.Thebatch procedure allowed only a limited number of carboxyl groups to effectively bondto the composite nanoparticles.In contrast,the number of carboxyl groups could bealtered over a wide range with the two-stage procedure.Fourier transform infraredspectroscopy and chemical titration were independently used to quantify the numberof carboxyl groups,giving values close to each other and to the feed.A kinetic studyindicated that the copolymerization followed a mechanism different than that foundearlier.The average size of the composite nanoparticles was approximately40nm,asmeasured by both transmission electron microscopy(TEM)and laser scattering,andtheir polydispersity index was close to1,indicating a fairly narrow size distribution.TEM photographs of the composite nanoparticles showed a multilayered core–shell structure with one silica bead as the core and with poly(methacrylate acid)as the outmost shell.V C2005Wiley Periodicals,Inc.J Polym Sci Part A:Polym Chem43:2826–2835,2005Keywords:composite nanoparticles;core-shell polymers;emulsifier-free;emulsion;copolymerization;functionalization of polymersINTRODUCTIONPolymer/inorganic composite nanoparticles are of growing interest not least because of their extensive applications asfillers in polymer-based nanocomposites.1–3The advantages of such composite nanoparticles over pure inor-ganic nanoparticles are obvious:not only is the composite material strengthened by the inor-ganic moiety,but the compatibility between the filler and the matrix is improved by the polymer moiety.Such composite nanoparticles can be prepared mainly by the encapsulation or graft-ing of polymers onto the surface of inorganic nanoparticles.An enormous amount of work has been reported,involving various inorganic par-ticles such as silica,4–6calcium carbonate,7tita-nium oxide,8–10and alumina11(among them, silica is the most widely studied).Encapsulation can be roughly divided into two types,physical and chemical,according on whether there is any chemical bonding at the interface between the inorganic nanoparticles andCorrespondence to:J.Yu(E-mail:yujian03@mail. )Journal of Polymer Science:Part A:Polymer Chemistry,Vol.43,2826–2835(2005) V C2005Wiley Periodicals,Inc.2826the polymer.Although physical encapsulation is simple,12,13chemical encapsulation is more inter-esting because of the stronger interfacial interac-tion provided by the covalent bonding at the inter-face.14,15There are mainly two approaches to achieve chemical encapsulation.One involves the use of inorganic nanoparticles pretreated with a silane coupling agent bearing a polymerizable dou-ble bond as a comonomer during the polymeriza-tion of vinyl monomers.16,17The other uses pre-treated silica as a macroinitiator for living polymerization.6,18,19For the former,the main polymerization techniques include emulsion and dispersion polymerization.Covalent bonds are formed between nanoparticles and polymer chains during encapsulation.For the latter,polymeriza-tion starts from the surface of inorganic nanopar-ticles,and encapsulation is formed as polymeriza-tion proceeds.As for the grafting of polymers onto inorganic nanoparticles,there are three main approaches that depend on the order of polymerization and grafting:grafting-from,20grafting-through,21and grafting-onto.22With the grafting-from approach, pretreated inorganic particles bearing initiating groups such as azo groups are used as initiators, and consequently polymerization starts from the surface of inorganic particles.The grafting-through approach uses pretreated inorganic nano-particles as comonomers of vinyl monomers.Graft-ing is achieved during polymerization.It may not necessarily form encapsulation;this depends on the polymerization conditions.As for the grafting-onto approach,prepolymers containing reactive groups are attached to inorganic particles.Although composite nanoparticles often show better dispersion and compatibility when used asfillers for polymers,the interfacial interaction between the composite nanoparticles and the polymer matrix is based only on physical com-patibility offered by the polymer shell and can certainly be improved further by the formation of chemical bonds,just like in the case of reac-tive blending.23For this purpose,composite nanoparticles bearing functional groups such as carboxyl groups(called reactivefillers)are needed and can potentially be useful in reinforc-ing polymers having functional groups that can react with the functional groups of thefiller.As part of an ongoing project aimed at the prep-aration of reactivefillers,we synthesized epoxy-functionalized polystyrene/silica composite nano-particles with a core–shell structure via emulsion polymerization.24A mixture of ionic and nonionicemulsifiers was used to stabilize the particles, which could partly remain in the latices as impur-ities(it is known that the emulsifiers used in emulsion polymerization are very hard to remove completely)and consequently limit the applica-tions of the latices to some degree.Emulsifier-free emulsion polymerization has been receiving considerably more attention in recent years because it can produce clean and monodisperse latices.25–28The emulsifier-free systems are often not truly free of an emulsifier in the strictest sense as the name indicates.The monomer or comonomer usually contains a part that resembles the structure of an emulsifier at one end of the molecular chain.Such a monomer or comonomer can play the role of an emulsifier while polymerizing.29Sodium methacrylate (NaMA)is one such comonomer.It is an ionic vinyl monomer with sodium carboxylate salt at one end of the molecule and a double bond at the other end,and it has been used to conduct emulsifier-free emulsion copolymerization.30 In this study,the emulsifier-free emulsion copolymerization of methyl methacrylate(MMA) was carried out to prepare reactive polymer/silica composite nanoparticles with NaMA as a comono-mer.Carboxyl groups were generated after the copolymerization by neutralization with hydro-chloric acid(HCl);this made the composite nano-particles reactive.With their interesting charac-teristics(clean surface,reactive and ionizable properties,and core–shell structure),such func-tional composite nanoparticles are expected to be used not only as reactivefillers but also in a wide range of applications such as protein carriers, microcapsules,water purifiers,and polymer cata-lysts.The method and amount of NaMA addition, the copolymerization kinetics,the carboxyl con-tent,and the morphology of the composite nano-particles are investigated here in detail. EXPERIMENTALMaterialsNanometer silica1065nm in diameter was acquired from Zhoushan Mingri Nanomaterial, Ltd.(China),and used after drying in vacuo at 1058C for12h.The pretreatment of silica with 3-methacryloxypropyltrimethoxysilane(MPTMS) was carried out with a previously published pro-cedure.24The monomers,both MMA and metha-crylate acid(MAA),were distilled under reduced pressure before use.NaMA was obtained by the POLYMER/SILICA COMPOSITE NANOPARTICLES2827neutralization of MAA with an equal molar amount of NaOH at08C.Ammonium persulfate (APS)was freshly recrystallized from water.All other reagents were used as received. CopolymerizationThe emulsifier-free emulsion copolymerization was carried out under a nitrogen atmosphere in a250-mL,four-neckedflask with a mechanical stirrer,thermometer,and condenser.For the whole process,the reaction was in a water bath, and the stirring rate wasfixed at150rpm. Batch ProcedureNaMA was introduced into a reactor charged with distilled water at408C.After20min of stirring at408C,the temperature was raised to 508C,and then a mixture of SiO2and MMA, which was treated by ultrasonic irradiation for 10min just before it was used,was added.After 10min of stirring at508C,the system was raised to608C,and a solution of APS in water was added to initiate the copolymerization.The reaction proceeded at808C for2h,and then it was raised to and held at908C for30min more. Excessive HCl was added to translate the acryl-ate into carboxyl groups;this also led to demul-sification.The product was washed thoroughly with hot water and then dried.Two-Stage ProcedureThefirst stage was similar to the batch proce-dure.After1h of stirring at808C,the second stage of copolymerization was started by the feeding of aqueous NaMA(at a rate of10mL/h)dropwise to the system.After the completion of aqueous NaMA addition,it was stirred at808C for30min and then was raised to and held at 908C for30min more.Excessive HCl was added to translate the acrylate into carboxyl groups; this also led to demulsification.The recipes of all runs are listed in Table1.The yield and conversion of the monomers were deter-mined by the gravimetric method as follows: Yieldð%Þ¼Total product(g)Total monomer(g)and SiO2ðgÞÂ100 Conversionð%Þ¼Polymer former(g)Monomer used(g)Â100To estimate the strength of the interaction between either PMMA and silica or PMMA and poly(methacrylate acid)(PMAA),the sample was extracted with chloroform for12h with a Soxh-let apparatus,and then the binding efficiency was calculated as follows:Binding efficiency(%)¼Polymer grafted(g)Polymer formed(g)Â100CharacterizationA latex sample was used directly for the mor-phology observation and particle size and size distribution determination with a JEOL200CX transmission electron microscope(dyed by RuO4 for20min)or a Zetaparticle HS3000laser scat-tering(LS)particle size and z-potential analyzer. Na was used to denote the number of silica beads per particle,and it was be calculated with a formula reported by Bourgeat-Lami andTable1.Recipes of Emulsifier-Free Emulsion CopolymerizationRun No.NaMA(mmol)MMA(mL)APS(mmol)H2O(mL)HCI(mmol) Portion1Portion2A a B bBatch series B1 2.1—15.00.2850.0— 5.0 B2 2.4—15.00.2850.0— 5.0B3 2.7—15.00.2850.0— 5.0B4 3.0—15.00.2850.0— 6.0B5 3.3—15.00.2850.0— 6.0 Two-stage series T1 2.7 5.515.00.2850.0 5.010.0 T2 2.713.715.00.2850.0 5.020.0T3 2.730.215.00.2850.010.050.0T4 2.746.715.00.2850.015.0100.0T5 2.763.215.00.2850.020.0100.0a Added before thefirst part of NaMA was introduced.b Used to dissolve the second part of NaMA.2828ZENG,YU,AND GUOLang.31The core–shell structure of composite nanoparticles can be considered well-defined,that is,only one silica bead per particle,when the value of Na ranges from 0.95to 1.05.The number-average diameter (D n )and the weight-average diameter (D w )were calculated with the following equations:D n ¼XiN i D i .X iN i :D w ¼XiN i D 4i.XiN i D 3i :where N i (i ¼1,2,...)is the number of the par-ticles with the size of D i (i ¼1,2,...).Both N i andD i are given by LS measurement.At least 105par-ticles (i.e.,Pi N i ,measured by LS)were counted for each calculation.The polydispersity index (PDI)of the particle size was expressed as D w /D n .A value ranging from 1.00to 1.05can be regarded as a monodisperse distribution of the particle size.Determination of the Contents of Carboxyl Groups The contents of carboxyl groups in the final car-boxyl-functional composite nanoparticles were determined by both Fourier transform infrared (FTIR)and chemical titration methods.In the for-mer,products bearing various contents of carboxyl groups after Soxhlet extraction were analyzed by FTIR with a Nicolet 560FTIR spectrometer.The vibration peak of the carbonyl group (1738cm À1)was taken as the reference peak.Thus,the area ratio of the carboxyl peak (1703cm À1)to the refer-ence peak could be calculated.The quantifications of the carboxyl contents (with respect to PMMA)were determined,with a calibration curve obtained from mixtures of MAA and MMA with different ratios.In the latter (i.e.,the titration method),a sample containing a product with a known mass was swollen in 1,4-dioxane for 24h.Then,a standardized NaOH solution with a known volume was introduced to neutralize the carboxyl group of the sample.The excess NaOH was then titrated by a standardized solution of an HCl–dioxane reagent with cresol red as an indica-tor.Experimental error due to dissolved CO 2was minimized by the performance of the titration under a nitrogen atmosphere.The difference between the number of moles of NaOH originally added and that neutralized by HCl equaled the number of moles of carboxyl groups.32Data from a minimum of three sets of these analyses were averaged.RESULTS AND DISCUSSIONBatch ProcedureIn this emulsifier-free emulsion copolymeriza-tion system,the hydrophilic ionic acrylate end and the hydrophobic carbon–carbon double-bond end made NaMA amphiphilic,so the role of NaMA was twofold:emulsifier and comonomer.Because silica was pretreated with MPTMS,it was hydrophobic and could be well dispersed in MMA after an ultrasonic treatment.WhenaScheme 1.Preparation of polymer/silica composite nanoparticles bearing carboxyl groups on the surface.POLYMER/SILICA COMPOSITE NANOPARTICLES 2829mixture of MPTMS-treated silica and MMA was introduced into the system containing the emul-sifier(NaMA),it broke and formed small drop-lets under the drive of emulsification and shear, and MMA absorbed onto the hydrophobic sur-face of MPTMS-treated silica.As an emulsifier, NaMA molecules covered the surfaces of the droplets and stabilized them.The hydrophilic ionic acrylate ends made NaMA molecules approach a water phase rather than mix with MMA homogeneously and go inside the droplets. Meanwhile,the hydrophobic double-bond ends admixed with the oil droplets and were oriented toward the center of the droplets,which could copolymerize with MMA and act as a comono-mer(Scheme1).Our previous work on the encapsulation of MPTMS-treated silica by poly-mers via emulsion polymerization has shown that the amount of the emulsifier is very impor-tant with respect to the binding efficiency.24 When excess emulsifiers were used,free latices formed,and this led to decreased binding effi-ciency.When the amount of the emulsifiers was not sufficient,partial demulsification easily hap-pened,especially in the presence of polar como-nomers,and this also led to decreased binding efficiency.Therefore,a series of copolymeriza-tions with different amounts of NaMA were per-formed,as shown in Table1.Figure1shows the effects of the amount of NaMA on the yield and binding efficiency.The trends of these two parameters are similar to those observed in our previous work mentioned previously in which sodium dodecyl sulfonate (SDS)was used as the emulsifier.It is clear that the optimal amount of NaMA was around 2.7mmol,at which both the yield and binding efficiency were higher than90%.With either less or more NaMA,the binding efficiency decreased, and partial demulsification or the formation of free latices occurred,respectively.It seems that NaMA behaves like the normal emulsifier SDS.The average size(D n)and size distribution (PDI)of the composite nanoparticles obtained with different recipes are listed in Table2.With an increasing amount of NaMA,the average particle size decreased gradually.Again,NaMA showed a typical behavior of a normal emulsi-fier.Only when the amount of NaMA was in the range of the optimal value did the obtained com-posite nanoparticles have a narrow size distribu-tion,and they could be considered monodisperse because the PDI was equal to1.04.With less or more NaMA,either the agglomeration of par-ticles(due to less emulsifier)or the formation of free latices without a silica core widened the size distribution.Two-Stage ProcedureAs mentioned previously,in the batch procedure, only with the optimal amount of NaMA feeding could the composite materials be obtained with a high yield and binding efficiency.Therefore, the number of functional groups that could be effectively bound to the composite nanoparticles was limited.To obtain composite nanoparticles with a wide range of functional groups,a two-stage procedure in view of NaMA addition was investigated.In thefirst stage,an optimal amount of NaMA was copolymerized with MMA (as recipe B3)so that latex seeds could be formed in a high yield and binding efficiency.In the second stage,NaMA was added dropwise to the system to keep NaMA under starved condi-tions to avoid the formation of freelatices.Table2.Evaluation of the Encapsulation withDifferent RecipesRecipe D n(nm)PDI NaB164.6 1.13—B253.8 1.14—B338.5 1.04 1.08B437.4 1.11—B535.2 1.17—T139.6 1.04 1.14T237.9 1.030.95T340.1 1.07 1.04T441.5 1.05 1.06T540.8 1.080.94 2830ZENG,YU,AND GUOFigure2shows the yield and binding effi-ciency versus the total amount of NaMA feed-ing.As expected,good encapsulation was obtained when the amount of NaMA varied over a wide range(from10to70mmol).The yields were all higher than90%,and the binding effi-ciencies were superior to85%.The high effi-ciency can be explained by the following points. First,the latex seeds obtained during thefirst stage had a very high binding efficiency(95%). Second,when NaMA was added to the emulsion system during the second stage,thefirst-stage copolymerization of MMA and MAA had justfin-ished,as indicated by the kinetic curve(shown later in Fig.5),the terminal free radicals of the copolymer chains were still alive,and the newly added NaMA could still copolymerize to the existing copolymer chains.Third,NaMA added during the second stage was adsorbed onto the latex particle and copolymerized almost instan-taneously because it was kept under starved conditions.33There was no formation of extra micelles,and this prevented the formation of free latices[pure poly(sodium methacrylate) (PNaMA)].Fourth,as the copolymerization of NaMA proceeded,the particles grew,the surfa-ces of the particles became larger,and more NaMA could be adsorbed and accommodated on the surface without saturation ever being reached.As shown in Table2,the average particle sizes were all around40nm with different amounts of NaMA feeding,and the particle size distributions were all narrow.In this two-stage procedure,the latex seed that formed during the first stage had a fairly narrow particle size dis-tribution(PDI¼1.04).During the second stage, the polymerization of NaMA occurred on the surface of the seeds,and little new latices formed because NaMA was kept under starved conditions.Therefore,thefinal composite par-ticles had a narrow size distribution. Determination of Carboxyl GroupsThe carboxyl contents of the composite nanopar-ticles were quantified independently with two methods:a physical method(FTIR)and a chemi-cal method(titration).Figure3shows the FTIR spectra of the products with different recipes. The band at1703cmÀ1in the spectra can be attributed to the C¼¼O stretching vibration of the carboxyl group,proving that the carboxyl group was bound onto the composite particles. In comparison with the spectrum of PMMA/ silica composite nanoparticles prepared previ-ously,the extra characteristic peaks of PNaMA at1566cmÀ1can hardly be observed,and this suggests that almost all of the salt was trans-lated into carboxyl groups.The intensity of the C¼¼O band increased with an increasing amount of NaMA feeding.The titration method was calibrated with known concentrations of acrylic acid,acetic acid, and HCl.As shown in Figure4,the average error in a set of(at least three)duplicate analy-ses for one compound at one carboxyl concentra-tion was63.2%.The correlation between the numbers of moles of carboxyl groups initially in each sample and that measured was linear with a slope of1.0027and an intercept of3.2Â10À5. Figure3.FTIR spectra of composite nanoparticles with different MAA feed concentrations:(a)0,(b)10, (c)20,(d)30,and(e)40%.Therefore,we can conclude that the titration method provided an accurate measurement of the number of carboxyl groups in our test samples.The results obtained with the two methods are listed in Table 3.The values obtained by the two different methods are close to each other for almost all the samples and show only small deviations from the amounts of added NaMA.For example,in recipe T3,when the amount of added NaMA was 23.2%(mol %to MMA),that is,the theoretical amount of carboxyl groups was 23.2%(mol %to MMA),the value quanti-fied by FTIR was 22.9%,and that measured by titration was 21.8%.The content of carboxyl groups increased with an increasing amount of NaMA feeding;this indicated that the content of carboxyl groups in the composite nanoparticles could be controlled by the feed.KineticsFigure 5shows the kinetic curves,with recipe T2as an example,with respect to the conver-sion and binding efficiency versus time.The sec-ond part of NaMA was fed dropwise into the sys-tem during the period marked with broken lines.The conversion–time curve is similar to that of the control experiment in the absence of silica (not shown),and this suggests that silica had almost no effect on the copolymerization kinetics.The conversion of the monomers (MMA and NaMA)increased rapidly during the initial 30min and then leveled off around 95%at about 50min.The copolymerization of the monomers added in the first stage was almost completed before the start of the second-stage addition of NaMA,which was carried out at 60min.During the dropwise addition of NaMA in the second stage,the conversion was almost unchanged (remaining at ca.95%),and this sug-gested that NaMA was kept under starved con-ditions.Because NaMA played the role of a normal emulsifier in the first stage,the curve of the binding efficiency versus the time in this period was the opposite of that of Espiard and Guyot,5who carried out the encapsulation of silica through the emulsion polymerization of ethyl acrylate (EA).In comparison with their system,the main differences in our case were the method of silica addition and the amount of the emulsifier.In their system,an aqueous silica solution was charged to the reactor before the addition of any other ingredients,and the mono-mer was fed dropwise after the initiator was introduced.However,the mixture of silica and MMA was added together in our case,andMMATable 3.Contents of Grafted PMAA Determined by Two MethodsRecipe Theoretical Amount of COOH (mol %to MMA)Actual Amount of COOH(mol %to PMMA)FTIR Titration B3 1.9 1.1 1.6T1 5.8 4.6 5.3T211.611.210.5T323.222.921.8T434.930.133.4T546.543.740.22832ZENG,YU,AND GUOwas well adsorbed onto the surface of MPTMS-treated silica before it was introduced to the emulsion system.Besides,a relatively large amount of the emulsifier(NaMA in thefirst stage)was used in our case,whereas the concen-tration of the emulsifier in their system was much lower than that of ours.We did an experi-ment with a procedure similar to that reported by Espiard and Guyot while keeping other parameters unchanged,and the binding effi-ciency of the product was only11%,much lower than that obtained with our procedure(ca. 90%).Another experiment was also carried out with much less emulsifier(0.2mmol of NaMA) and with other parameters unchanged;it resulted in only15%binding efficiency,which agreed with the trend shown by Figure1.The results of these two experiments emphasize the importance of both parameters,that is,the method of silica addition and the amount of the emulsifier(NaMA in thefirst stage).In our case,as mentioned in the Batch Proce-dure section,hydrophobic MPTMS-treated silica dispersed well in MMA,and then the mixture was introduced into the system.Under the drive of emulsification and shear,the mixture was separated into small droplets of MMA contain-ing MPTMS-treated rger pure MMA droplets hardly existed because if they had existed,they would have broken and formed smaller pure MMA droplets,which would have polymerized and formed free polymer latices finally.Because the binding efficiency was nearly100%(shown in Fig.5),there was almost no free polymer latex.Therefore,after the free radicals of the initiator came into the droplets, copolymerization may have taken place in the droplets containing the MPTMS-treated silica particles,which were surrounded by MMA,as shown in Scheme 1.Polymers were formed around the surface of silica and were covalently bound to silica when silica reacted as a comono-mer.The binding efficiency during the initial 15min was very low,whereas the conversion of the monomers increased rapidly during this period.This suggests that the polymer that formed during the initial15min hardly bonded to silica;that is,the double bonds of the MPTMS-treated silica practically did not react. It is probable that the initiator radicals entered the silica/MMA/NaMA droplets from the outside and initiated preferentially the monomers (MMA and NaMA)that were near the surfaces of the droplets(Scheme1).As the copolymeriza-tion proceeded,the growing radicals had more chance to reach the center of the droplets and react with the double bonds of silica,forming grafting,and the binding efficiency increased accordingly.Moreover,the binding efficiency achieved its maximum at about40min and then leveled off,whereas the monomer conversion was still increasing at that time.It can be con-cluded that the reaction of the double bonds on the surface of silica was almost completed after 40min of copolymerization.The monomers con-tinued polymerizing at the polymer shell and propagated onto the polymer chains(rather than form new polymer chains)after the complete reaction of silica;this was similar to the shell growth mechanism presented by Yan et al.34 Therefore,the reaction of silica in our case mainly happened from15to40min,and so the binding efficiency increased rapidly after15min of reaction and reached its maximum at about 40min of reaction.In the case of Espiard and Guyot,5MPTMS-treated silica was added in the beginning,and the initiator was added before the dropwise addition of the monomer.Hence,the double bonds of MPTMS-treated silica had a greater chance to be initiated during the early stage of polymerization,and grafting could start from the surface of silica.As the polymerization pro-ceeded,more emulsifier was needed tostabilize Scheme2.Polymerization mechanism of Espiard and Guyot’s system[PEA¼poly(ethyl acrylate)].POLYMER/SILICA COMPOSITE NANOPARTICLES2833the growing latices.However,the amount of the emulsifier was extremely small,and the mono-mer added in the later stage tended to form pure polymer latex,following the homogeneous nucleation mechanism of emulsifier-free emul-sion polymerization;this resulted in decreased grafting efficiency,as shown in Scheme 2.MorphologyTransmission electron microscopy (TEM)photo-graphs of primary silica beads and carboxyl-functionalized composite nanoparticles taken from the emulsion after copolymerization was fin-ished and dyed by RuO 4are shown in Figure 6.The multilayered core–shell structure can be ob-served in the image of the final particles [Fig.6(b)],which is different from that of the primary silica beads [Fig.6(a)].In each composite nanopar-ticle,the dark core is made of only one silica bead;this is proved by both its size and the cal-culation of Na (as shown in Table 2)and sug-gests the formation of a well-defined core–shell structure.The surrounding light layer is PMMA,which cannot be dyed by RuO 4.The out-most dark shell is PMAA containing carboxyl groups,which is dyed by RuO 4.This kind of par-ticle morphology is in agreement with our expectations.The average particle size deter-mined by TEM was about 40nm with a narrow size distribution,which agreed with that meas-ured by LS (as shown in Table 2).CONCLUSIONSThe emulsifier-free emulsion copolymerization of MMA and NaMA has been successfully used to prepare functional polymer/silica composite nano-particles.It has been shown that the method of NaMA feeding is very important with respect to the yield and binding efficiency,and only the two-stage procedure allows effective control of the carboxyl content,which was measured by two different methods (FTIR and titration).The kinetic study suggests that the copolymerization follows a mechanism different than that found earlier.Polymers formed during the initial stage practically do not graft onto silica,and the mono-mers continue polymerizing,following the shell growth mechanism,after silica has completely reacted;therefore,the binding efficiency increases rapidly after 15min and reaches its maximum before the copolymerization finishes.As demon-strated by TEM,the composite nanoparticles have a multilayered core–shell structure with silica as the core and with PMAA as the outmost shell.The particles sizes are around 40nm with a narrow size distribution.REFERENCES AND NOTES1.Chen,J.F.;Wang,G.Q.;Zeng,X.F.;Zhao,H.Y.;Cao,D.P .;Yun,J.;Tan,C.K.J Appl Polym Sci 2004,94,796–802.Figure 6.TEM photographs of (a)primary silica beads and (b)carboxyl-functionalized composite nanoparticles.2834ZENG,YU,AND GUO。
化学交联聚乙烯醇改性纤维素碱性阴离子交换复合膜的制备与性能
关键 词: 碱 性阴离子交换膜: 聚 乙烯醇; 季铵化羟 乙基 乙氧基纤维素: 共混化学交联 : OH 电导率 一
耐 碱 稳 定 性
中图分类号 : 06 6 4
Syn h s s a op te f t e i nd Pr eri s o e i a l o s Li k d Ch m c l Cr s - n e y
P l(iy lo o) df dQu tr e y rx eh IelIs oyvn l c h 1Mo ie ae i dH d o y tyc I o e a i z u
Et ox a e a ve k l e An on E h n h yl t s No l Al a i i ・ xc a ge Mem b an n r e
me rn swee c aa tr e sn o r rt n fr nrrd (T R p cr ,h r ga i tc(G) mba e r h rcei d u ig F u i r som i ae F I )s e t temo rvmer T z e a f a i
Ab t a t M u h a t n i a e n p i o a k l e i . x h g mbr e t i h a k i e s a it . n sr c: c t t e on h s b e a d t la i n on e c an e me an s wi h g lal t b ly I h n i
( , QHECE)me r n s we e p e a e n v lae s OH—c n u t g p lme lcr lt s h P、 A, mb a e r r p r d a d e au td a o d c i oy ree toye .T e n
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
第2卷第4期2003年10月 江南大学学报(自然科学版)JournalofSouthernYangtzeUniversity(NaturalScienceEdition)
Vol.2 No.4
Oct. 2003
文章编号:1671-7147(2003)04-0406-03
收稿日期:2003-02-19; 修订日期:2003-04-10.
作者简介:沈晓东(1964-),男,江苏无锡人,工学硕士,副教授.
聚乙烯的紫外交联沈晓东, 龚雁(江南大学化学与材料工程学院,江苏无锡214036)
摘 要:利用2,32二甲基22,32二苯基丁烷作为自由基引发剂,用紫外光照射引发聚乙烯的交联.
讨论了引发剂的用量以及紫外光照射时间对聚乙烯交联程度的影响,并与过氧化二异丙苯进行比较试验,结果证明了2,32二甲基22,32二苯基丁烷具有较强的引发交联能力.
关键词:2,32二甲基22,32二苯基丁烷;自由基;聚乙烯;交联中图分类号:TQ314.241文献标识码:A
UltravioletCrosslinkingofPolyethyleneSHENXiao2dong, GONGYan(SchoolofChemicalandMaterialEngineering,SouthernYangtzeUniversity,Wuxi214036,China)
Abstract:2,32dimethyl22,32diphenylbutancewasusedasfreeradicalinitiatortopreparecrosslinkedpolyethylenebyultravioletlampsirradiation.VariousquantityofDMDPBandultravioletlightirradiationtimewerestudiedabouttheirinfluenceofcrosslinkingdegreeofpolyethylene.DicumylperoxidewascomparedwithDMDPB.TheresultshowedthatDMDPBhasstrongercapabilityofinitiationcrosslinking.Keywords:2,32dimethyl22,32diphenylbutance;freeradicalpolyethylene;crosslinking
交联共聚是高分子材料改善性能的一条重要途径,通过交联这一途径常可获得综合性能较为理想的高分子材料.聚烯烃广泛应用于电力电缆和通讯电缆的绝缘材料,但耐热性不佳.交联后可提高聚烯烃在高温下的耐热应变能力及作为绝缘体的使用温度,增加应用的安全性.聚烯烃的交联主要采用化学交联法和辐射交联法[1].化学交联法一般采用有机过氧化物作为交联剂,通过加热使有机过氧化物分解而引发交联;辐射交联法则用各种高能射线(如γ射线)辐照使之交联[2].化学交联法所用的有机过氧化物一般引发温度较低,由于聚烯烃(如聚乙烯)的混和、加工、成型温度较高,在此条件下,有机过氧化物可能提前发生分解而使聚乙烯的可塑性下降.辐射交联法使用高能射线,对设备要求高,应用范围受到一定限制.
文中通过在聚烯烃中加入2,32二甲基22,32二苯基丁烷(DMDPB)而引发交联.DMDPB是一种C—C型自由基引发剂[3],与过氧化物类自由基引发剂相比,DMDPB形成的自由基活性大,夺氢能力强,可通过在聚烯烃的碳链上生成光交联点[4]使聚烯烃形成交联结构.国外有用于耐热绝缘电缆[5]及耐热聚合物[6]制造的相关报道.自由基的引发方式有2种,一种是加热引发,另一种可通过各种射线辐照引发.经DSC测定DMDPB的引发温度在200
℃以上,296℃达到分解的最大值.这一引发温度比一般过氧化物的引发温度高100℃左右,远高于一般塑料的加工成型温度.这在加工过程中可避免因引发剂的过早分解而导致塑料的可塑性下降,但200℃以上的温度对塑料的加工成型可能造成不利影响,因此加热引发DMDPB分解的方式在交联或接枝应用时受到限制.在实验中,作者采用了紫外光照射的方式使DMDPB分解而引发交联.交联对高聚物的结构和物理性质会产生影响,通过对某些性质的测定可以表征高聚物的交联程度[7].文中以聚乙烯在甲苯中的溶解性表示其交联程度:交联后的聚乙烯不溶于甲苯,溶解后的残留量(凝胶含量)与交联度成正比.1 材料与方法1.1 试剂与设备1.1.1 试剂1聚乙烯,2F2B,齐鲁石化股份有限公司生产;过氧化二异丙苯(DCP),化学纯,上海化学试剂站中心化工厂生产;2,32二甲基22,32二苯基丁烷(DMDPB),自制,熔点为116~118℃.1.1.2 仪器1SK2160B型双辊筒炼塑机,上海橡胶机械厂制造;平板硫化机,上海轻工机械有限公司制造;紫外线照射仪,涿洲市蓝天光源设备厂制造.1.2 试样的制备按配方要求准确计量,将各组分按一定顺序混合后,在125℃条件下,用双辊筒炼塑机进行混炼.分散均匀后,控制一定的温度,在平板硫化机上成型,用紫外线照射仪对成型后的样品照射一定的时间,得到试样.1.3 交联度的测定准确称取1g左右试样,用滤纸包裹,小心放入脂肪抽提器中,加入500mL甲苯,加热回流6h,取出试样烘干称其质量,根据残留量确定交联程度.2 结果与讨论2.1 加热与紫外光照射方式的交联结果比较两种不同引发方式的交联结果如图1所示[8]. 从交联结果可以看出,在引发剂用量相同的情况下,由紫外光所引发的交联程度比加热高.在加热实验中所使用的温度为300℃,远超过一般塑料的加工温度,但引发的效果仍不理想,且加热后的聚乙烯颜色变黄,可塑性明显下降,说明加热引发的方法不适宜实际应用.由紫外光所引发的交联程度较高,在不加引发剂的条件下也会引起聚乙烯的部分交联,但程度较小(1%~3%).加入引发剂DMDPB后,交联程度迅速增大,并随着DMDPB用量的增加而增大,但DMDPB用量增加与交联程度不成正比关系.这与DMDPB的分解速度和交联速度以及其它因素(如DMDPB的分散程度、自由基的效率、聚乙烯的交联密度等)有关.当反应开始时,
DMDPB的含量较高,分解速度快,引发交联的程度深;DMDPB的分散度高,交联度大;对聚乙烯的交联密度而言,当链与链之间的交联形成后,再形成交联键则对交联度没有贡献.当加入DMDPB的质量分数为2%时,即有较高的交联度.
注:加热温度为300℃,时间为8min,紫外线照射时间为20min.
图1 加热与紫外交联结果比较Fig.1 Comparisonofcrosslinkingresultheatingandultravioletirradiation2.2 照射时间对交联程度的影响紫外光照射时间对交联程度的影响见图2.
图2 紫外光照射时间的影响Fig.2 Theinfluenceofultravioletlightirradiationtime DMDPB的分解由紫外光所引发,照射时间增加,DMDPB的分解率增大,所引发的交联程度也逐渐增加.但从增加的幅度看,随着照射时间的增加,
交联程度增加的幅度逐渐变小.随着反应的进行,
DMDPB的含量逐渐减少,由此引发的交联程度变小.且照射时间长,温度升高,引发其他反应,使聚合物的性能发生变化.
2.3 DMDPB与DCP的比较分别把DMDPB,DCP与聚乙烯混和后制成样品,加入质量分数为2%的聚乙烯.经紫外光照射20
min后,测定残留量,确定其交联程度.结果如图3
704 第4期沈晓东:聚乙烯的紫外交联所示.图3 DMDPB与DCP的比较Fig.3 ComparisonofDMDPBandDCP DCP是一种常用的自由基引发剂,它的引发温度较低,大于120℃即可分解且形成自由基,因而在与聚乙烯混塑的过程中已发生分解而引发交联,使塑料的硬度变大,熔点上升,可塑性下降.而DMDPB的分解温度高,因此在混炼过程中不会引发塑料的过早交联.紫外光的照射同样可使DCP分解引发交联,但随着照射时间的增加,引发程度变低,这是因为DCP的起始分解速度大,但所形成的自由基稳定性相对较高,夺氢能力相对较弱,交联程度较小.与DCP相比较,DMDPB在的紫外光的照射下所引发的交联程度要大,且交联程度可通过照射时间进行控制.
3 结 语在聚乙烯加入DMDPB后通过紫外光的照射可以引发聚合物的交联,加入量和照射时间对交联度有影响,DMDPB加入量的增加和照射时间的延长会加大交联程度.与DCP相比较,DMDPB在的紫外光的照射下引发聚合物交联的能力强,且在混炼过程中不会引发塑料的过早交联而使可加工性下降.
参考文献:
[1]许长清.合成树脂及塑料手册[M].北京:化学工业出版社,1991.28-29.[2]张志成,葛学武,张曼维.高分子辐射化学[M].合肥:中国科学技术大学出版社,2000.[3]沈晓东,丁先锋,王建新.新型的聚合物改性剂[J].江南学院学报,1999,14(3):85-88.[4]瞿保钧,徐云华,瞿欣.低密度聚乙烯光交联点的结构及其形成机理[J].高等学校化学学报,1997,18(2):317-322.[5]MAEDAKAZUNORI,NAGAIKENJI.Heat—resistantelectricallyinsulatingpolymercomposition[P].日本专利:JP07105735,1995208209.[6]AOKINOBUO,ENOMOTOTOSHIYUKI.Manufactureofthermosettingheat—resistantpolymerusingdialkylbutanes[P].欧洲专利:EP389076,1991205204.
[7]何曼君,陈维孝,董西侠.高分子物理[M].上海:复旦大学出版社,1991.[8]沈晓东,王建新.二甲基二苯基丁烷的应用[J].中国塑料,2000,83(2):65-68.(责任编辑:邢宝妹)
《无锡轻工大学学报》2004年征订启事《无锡轻工大学学报》(双月刊)是教育部主管、江南大学(原无锡轻工大学)主办的有关食品、生物工程及其相关研究的专业性学术期刊,为全国中文核心期刊,中国期刊方阵双效期刊,目前被美国化学文摘(CA)等国内外10余家著名检索系统收录。本刊主要刊发食品科学与工程,食品营养学,粮食、油脂及植物蛋白工程,制糖工程,农产品及水产品加工与贮藏,微生物发酵,生物制药工程等专业最新科研成果(新理论、新方法、新技术)的学术论文、试验报告、反映学科前沿研究动态的高质量综述文章,同时兼发动物营养与饲料工程、环境生物技术等方面的最新研究成果。本刊读者对象:相关领域的高等院校、科研院所、企事业单位的教学、科研等专业技术人员,专业管理人员以及有关院校师生。本刊热忱欢迎广大读者订阅。《无锡轻工大学学报》,A4(大16K)开本,112页,每册定价8.00元,全年6期48.00元;本刊邮发代号: