全基因组测序案例
水产基因组群体gwas案例

水产基因组群体gwas案例标题:水产基因组群体GWAS案例一、引言随着高通量测序技术的发展,水产基因组研究取得了巨大的进展。
基因组关联分析(Genome-Wide Association Study,GWAS)是一种常用的遗传学方法,可以帮助我们识别与性状相关的基因。
本文将以水产基因组群体GWAS案例为题,介绍一些相关的研究成果。
二、鲈鱼GWAS研究鲈鱼是一种重要的水产养殖品种,其肉质品质是影响市场价值的关键因素之一。
一项研究通过GWAS分析,发现了与鲈鱼肉质品质相关的基因突变。
这些基因突变可能与肌肉生长和脂肪代谢有关,为改良鲈鱼的肉质品质提供了潜在的遗传基础。
三、虾类GWAS研究虾类是另一类重要的水产养殖品种,其生长性能和抗病能力是养殖成功的关键。
一项GWAS研究发现,一些基因与虾类的生长性状和抗病性状密切相关。
例如,某个特定基因的变异可能影响虾类的体重增长速度,而另一个基因的变异可能增强虾类的抗病能力。
这些发现为虾类育种提供了重要的遗传信息。
四、鲑鱼GWAS研究鲑鱼是一种重要的商业养殖鱼类,其肉质品质和抗病能力是养殖者关注的重点。
一项GWAS研究发现,一些基因与鲑鱼的脂肪含量和抗病能力密切相关。
这些基因可能调控脂肪代谢途径和免疫反应,为改良鲑鱼的肉质品质和抗病能力提供了潜在的遗传基础。
五、蛤蜊GWAS研究蛤蜊是一种广泛分布的贝类,其耐受力和适应性是研究者关注的重点。
一项GWAS研究发现,一些基因与蛤蜊的耐受力和适应性密切相关。
例如,某个特定基因的变异可能增强蛤蜊对环境胁迫的适应能力,而另一个基因的变异可能增强蛤蜊对病原微生物的抵抗能力。
这些发现为蛤蜊育种和保护提供了重要的遗传信息。
六、鳕鱼GWAS研究鳕鱼是一种重要的渔业资源,其生长性能和抗病能力是研究者关注的焦点。
一项GWAS研究发现,一些基因与鳕鱼的生长性状和抗病性状密切相关。
这些基因可能影响鳕鱼的生长速度和免疫反应,为改良鳕鱼的生长性能和抗病能力提供了潜在的遗传基础。
‘申鸿七彩雉鸡’mtDNA 全基因组序列测序和系统发育关系分析

特产研究25Special Wild Economic Animal and Plant ResearchDOI:10.16720/ki.tcyj.2021.046‘申鸿七彩雉鸡’全基因组序列测序和系统发育关系分析陈芳1,覃健峰1,郑德滨1,袁红艳2,吴琼1※(1.龙岩学院生命科学学院,福建龙岩364012;2.上海欣灏珍禽育种有限公司,上海201400)摘要:为了从分子角度解析‘申鸿七彩雉鸡’种群遗传背景,本研究采用PCR测序方法,对3个个体‘申鸿七彩雉鸡’的线粒体全基因组进行测序,并与NCBI公布的7条雉鸡线粒体基因组序列比对,构建系统进化树。
获得‘申鸿七彩雉鸡’线粒体全基因组序列全长为16685bp,结构为典型的环状双链,包括13个蛋白质编码基因、2个rRNA、22个tRNA和D-Loop区。
蛋白编码基因总长度为11369bp,基因重叠有7处,间隔有20处;D-loop序列长为1148bp,占整个基因的6.88%;tRNA基因长度为65~78nt。
‘申鸿七彩雉鸡’线粒体全基因组、蛋白质编码基因、tRNA、rRNA和D-Loop区碱基组成中AT含量均大于CG含量。
构建的进化树发现,‘申鸿七彩雉鸡’和‘甘肃亚种雉鸡’聚为一类。
关键词:‘申鸿七彩雉鸡’;线粒体全基因组;系统发育关系中图分类号:S865.3文献标识码:A文章编号:1001-4721(2022)02-0025-06CHEN Fang1,QIN Jian-feng1,ZHENG De-bin1,YUAN Hong-yan2,WU Qiong1※(1.College of Life Science,Longyan University,Longyan364012,China;2.Shanghai Xinhao Zhen Poultry Breeding Co.,Ltd.,Shanghai201400,China):The aim of this study was to reveal the genetic relationship of'Shenhong pheasant'in China.The mitochondrial genome of three pheasant varieties were sequenced and analyzed to study the genetic diversity by PCR method.The results showed that the entire mtDNA sequence was16685bp in length and contained13protein-coding genes,2rRNA genes,22tRNA genes,and a control region(D-loop),Its gene arrangement pattern was identical with those of other avians.The total length of protein coding gene is11369bp,with7overlapping genes and20intervals.The length of D-loop sequence is1148bp,accounting for6.88%of the whole gene.The length of tRNA gene is 65-78nt.The AT content in the whole mitochondrial genome,protein coding genes,tRNA,rRNA and D-Loop regions of'Shenhong pheas-ant'are higher than CG content.'Phasianus colchicus strauchi'and'Shenhong pheasant'were closer by evolutionarytree.:'Shenhong pheasant';mitochondrial DNA;phylogenetic relationship雉鸡,学名环颈雉,俗称野鸡、山鸡。
全基因组重测序技术在牛品种鉴别与遗传多样性研究中的应用

全基因组重测序技术在牛品种鉴别与遗传多样性研究中的应用近年来,全基因组重测序技术在生物学领域中得到了广泛应用。
全基因组重测序技术能够高精度、高通量地测定一个组织的所有基因,包括编码区和非编码区,进而得到一个组织的全基因组序列。
与传统的SNP分析技术相比较,全基因组重测序技术具有更高的精度和更全面的分析能力。
在农业领域中,全基因组重测序技术不仅可以用于土壤微生物群体的多样性分析,还可以用于畜禽种群的鉴别和遗传多样性研究。
随着现代农业的发展,畜肉市场的重要性日益增加。
畜禽品种多样,不同品种的肉质和产量也有所不同。
针对这些问题,牛品种鉴别和遗传多样性研究变得十分重要。
全基因组重测序技术的兴起为这些问题提供了解决方案。
与传统的PCR和单倍型分析技术相比,全基因组重测序技术在牛品种鉴别和遗传多样性研究中具有更好的准确性和更高的效率。
在牛品种鉴别方面,全基因组重测序技术能够区分不同品种之间的基因大小和基因序列信息。
通过对多个牛品种进行全基因组重测序,可以得到大量的基因组数据,进而分析不同品种之间的遗传差异。
这种方法已被广泛用于不同牛品种的分析。
同时,这一技术还可以用于检测不同的基因突变,在品种鉴别和遗传多样性研究中发挥关键作用。
在遗传多样性研究方面,全基因组重测序技术可以通过分析单核苷酸多态性(SNP)来揭示一个物种的遗传多样性。
通过不同品种牛之间SNP差异的比较,可以推断它们之间的亲缘关系和遗传相似性。
这种方法基于遗传相异性和遗传距离的测量,能够更加准确地评估不同品种牛之间的相关性。
在遗传多样性研究中,通过全基因组重测序技术得到的SNP位点信息,可以用于构建不同品种之间的遗传距离矩阵,从而更好地评估不同品种之间的遗传多样性。
此外,全基因组重测序技术在畜牧业中的应用还包括了个体基因检测、基因编辑等方面。
这些研究的结果显著提高了畜禽育种的效率和生产力,进一步为畜肉市场的发展提供了有力支持和保障。
总之,全基因组重测序技术在牛品种鉴别和遗传多样性研究中的应用是十分重要的。
案例解读长读长测序技术成功鉴定神经元核内包涵体病(NIID)致病机制
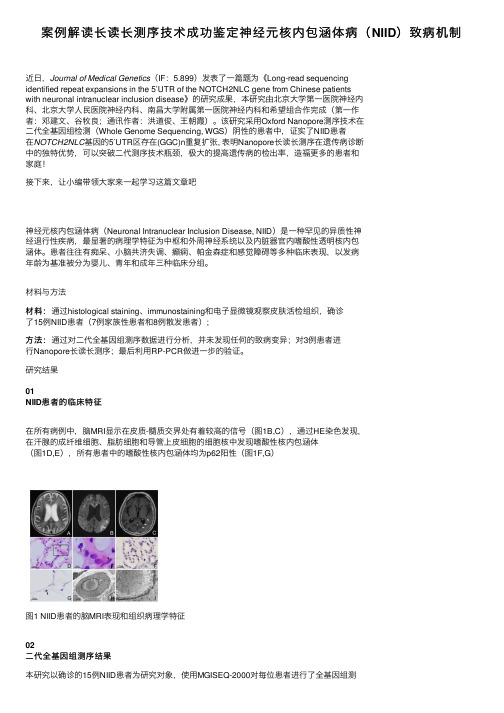
案例解读长读长测序技术成功鉴定神经元核内包涵体病(NIID)致病机制近⽇,Journal of Medical Genetics(IF:5.899)发表了⼀篇题为《Long-read sequencingidentified repeat expansions in the 5′UTR of the NOTCH2NLC gene from Chinese patientswith neuronal intranuclear inclusion disease》的研究成果,本研究由北京⼤学第⼀医院神经内科、北京⼤学⼈民医院神经内科、南昌⼤学附属第⼀医院神经内科和希望组合作完成(第⼀作者:邓建⽂、⾕牧良;通讯作者:洪道俊、王朝霞)。
该研究采⽤Oxford Nanopore测序技术在⼆代全基因组检测(Whole Genome Sequencing, WGS)阴性的患者中,证实了NIID患者在NOTCH2NLC基因的5′UTR区存在(GGC)n重复扩张, 表明Nanopore长读长测序在遗传病诊断中的独特优势,可以突破⼆代测序技术瓶颈,极⼤的提⾼遗传病的检出率,造福更多的患者和家庭!接下来,让⼩编带领⼤家来⼀起学习这篇⽂章吧神经元核内包涵体病(Neuronal Intranuclear Inclusion Disease, NIID)是⼀种罕见的异质性神经退⾏性疾病,最显著的病理学特征为中枢和外周神经系统以及内脏器官内嗜酸性透明核内包涵体。
患者往往有痴呆、⼩脑共济失调、癫痫、帕⾦森症和感觉障碍等多种临床表现,以发病年龄为基准被分为婴⼉、青年和成年三种临床分组。
材料与⽅法材料:通过histological staining、immunostaining和电⼦显微镜观察⽪肤活检组织,确诊材料:了15例NIID患者(7例家族性患者和8例散发患者);⽅法:通过对⼆代全基因组测序数据进⾏分析,并未发现任何的致病变异;对3例患者进⽅法:⾏Nanopore长读长测序;最后利⽤RP-PCR做进⼀步的验证。
贝纳基因 全基因组组测序报告解读

贝纳基因全基因组组测序报告解读全文共四篇示例,供读者参考第一篇示例:贝纳基因全基因组组测序报告解读贝纳基因是一家致力于基因检测与解读的公司,旨在帮助人们更好地了解自己的基因信息,为未来的健康生活提供科学指导。
全基因组组测序是一种高通量的基因检测技术,可以快速准确地分析个体的所有基因信息,帮助人们深入了解自己的遗传特征,预防潜在的疾病风险。
在接受全基因组组测序后,贝纳基因会提供详细的报告解读,帮助用户理解自己的基因信息,制定个性化的健康管理方案。
全基因组组测序报告是一份涵盖多个方面的综合性报告,包括基因组结构、基因型、基因突变、疾病风险等内容。
在解读报告时,用户需要了解一些基本的遗传学知识,以便更好地理解报告内容。
用户需要了解基因是DNA分子的一部分,携带着人类的遗传信息,影响个体的生理和疾病易感性。
基因组是指一个人的所有基因的总和,包括编码基因和非编码基因。
全基因组组测序技术可以对一个人的整个基因组进行测序,帮助用户了解自己的基因特征。
在报告解读过程中,用户可以了解自己的基因型,即基因的不同等位基因的组合。
基因型是由父母遗传给子代的,不同基因型可能影响个体的生理特征或疾病易感性。
报告中还会包括基因突变的信息,即基因序列发生变异或突变的现象。
基因突变可能导致某些疾病的发生,例如遗传疾病或癌症。
贝纳基因将会为用户提供关于基因突变的详细解读,帮助用户了解自己的疾病风险。
除了基因型和基因突变外,报告还会对用户的疾病风险进行分析和评估。
贝纳基因的报告会根据用户的基因信息,预测可能患上某些常见疾病的风险,例如心血管疾病、糖尿病、遗传疾病等。
用户可以根据报告中的建议,制定相应的健康管理方案,预防潜在的疾病风险。
报告还会为用户提供个性化的营养建议、运动计划、药物反应等方面的信息,帮助用户更好地管理自己的健康。
在阅读全基因组组测序报告时,用户需要对其中的信息进行理性分析和判断。
基因检测只是一个参考指标,不能完全代表个体的健康状况。
全基因组关联分析剖析

对家系数据进行检查,排 除样本混淆、亲子关系 错误等问题,控制家系关 系的正确性。
全基因组关联分析的结果验证
验证检查
对于全基因组关联分析的结果,需要进行严格的验证检查,以确保结果的可靠性和重复性。
重复实验
在不同的人群或样本中重复实验,比较结果是否一致进一步的功能实验,探讨基因变异与表型之间的机制。
全基因组关联分析的统计方法
统计分析
全基因组关联分析通常采用统计模型对遗传标记与表型之间的关联进行测试,如线性回归、logistic 回归等。
多重检验校正
由于基因组级别的大量比较检验,需要采用Bonferroni、FDR等方法进行多重检验校正,以控制I型错 误风险。
机器学习方法
近年来,全基因组关联分析也开始采用机器学习技术,如Ridge回归、Lasso回归等方法,以提高检测 能力。
全基因组关联分析的研究 热点
1 复杂疾病研究
全基因组关联分析被广 泛应用于探索复杂疾病 如糖尿病、心血管疾病 、肿瘤等的遗传学基础 。
3 交互作用研究
多基因、基因-环境等交 互作用的研究是全基因 组关联分析的重要方向 。
2 药物反应预测
全基因组分析有助于识 别影响药物反应的基因 变异,助力个体化精准医 疗。
生物学解释
从统计上显著关联的遗 传位点到生物学功能解 释存在鸿沟,需要更深入 的研究。
跨人群适用性
现有大多数研究集中于 欧美人群,如何推广到其 他人群是一大挑战。
全基因组关联分析的研究进 展
多组学整合
研究者正在探索将全基因组 关联分析与转录组学、表观 遗传学等多种组学数据相结 合的方法,以更全面地了解 复杂疾病的遗传学机制。
新型统计方法
学者们不断开发基于机器学 习、贝叶斯统计等的创新分 析方法,以提高检测复杂遗 传变异和基因-环境相互作 用的能力。
细菌重测序
Morrisania Central Bronx Bronx Park Riverdale
Outer circle: Isolate type
clinical colonizer enviromental
图1 致病菌株进化规律及其地域属性
[案例二] 不同测序平台在合成气发酵梭菌完成图构建中的比较[2]
Middle circle: Neighborhood
Inwood Fort George Washington Heights Hamilton Heights Harlem
East Harlem Hunts point, mott haven Houston, Texas San Diego, California San Francisco, California
[2] Brown S D, Nagaraju S, Utturkar S, et al. Comparison of single-molecule sequencing and hybrid approaches for finishing the genome of Clostridium autoethanogenum and analysis of CRISPR systems in industrial relevant Clostridia [J]. Biotechnol. Biofuels, 2014, 7: 40.
参考文献
图2 各平台对合成气发酵梭菌基因组装结果比较
[1] Uhlemann A C, Dordel J, Knox J R, et al. Molecular tracing of the emergence, diversification, and transmission of S. aureus sequence type 8 in a New York community [J]. Proceedings of the National Academy of Sciences, 2014, 111(18): 6738-6743.
全基因组重测序数据分析详细说明
全基因组重测序数据分析1. 简介(Introduction)通过高通量测序识别发现de novo的somatic和germ line 突变,结构变异-SNV,包括重排突变(deletioin, duplication 以及copy number variation)以及SNP的座位;针对重排突变和SNP的功能性进行综合分析;我们将分析基因功能(包括miRNA),重组率(Recombination)情况,杂合性缺失(LOH)以及进化选择与mutation之间的关系;以及这些关系将怎样使得在disease(cancer)genome中的mutation产生对应的易感机制和功能。
我们将在基因组学以及比较基因组学,群体遗传学综合层面上深入探索疾病基因组和癌症基因组。
实验设计与样本(1)Case-Control 对照组设计;(2)家庭成员组设计:父母-子女组(4人、3人组或多人);初级数据分析1.数据量产出:总碱基数量、Total Mapping Reads、Uniquely Mapping Reads统计,测序深度分析。
2.一致性序列组装:与参考基因组序列(Reference genome sequence)的比对分析,利用贝叶斯统计模型检测出每个碱基位点的最大可能性基因型,并组装出该个体基因组的一致序列。
3.SNP检测及在基因组中的分布:提取全基因组中所有多态性位点,结合质量值、测序深度、重复性等因素作进一步的过滤筛选,最终得到可信度高的SNP数据集。
并根据参考基因组信息对检测到的变异进行注释。
4.InDel检测及在基因组的分布: 在进行mapping的过程中,进行容gap的比对并检测可信的short InDel。
在检测过程中,gap的长度为1~5个碱基。
对于每个InDel的检测,至少需要3个Paired-End序列的支持。
5.Structure Variation检测及在基因组中的分布: 能够检测到的结构变异类型主要有:插入、缺失、复制、倒位、易位等。
基因组结构变异分析的常用方法与实例
基因组结构变异分析的常用方法与实例基因组结构变异是指DNA序列的改变,包括插入、缺失、倒位、复制和转座等不同类型的变异。
这些变异对生物的产生、进化、发育和疾病等多个方面具有重要影响。
因此,研究基因组结构变异对于理解生物遗传学和进化生物学等领域具有重要意义。
本篇文章将介绍基因组结构变异分析的常用方法以及一些实例。
1. 数字比对方法数字比对方法是通过使用计算机算法将测序数据与参考基因组进行比对,以检测基因组结构变异。
其中,最常用的算法是BLAST(Basic Local Alignment Search Tool)和BWA(Burrows-Wheeler Aligner)。
这些算法能够高效地识别出插入、缺失和倒位等结构变异。
2. 高通量测序方法高通量测序方法是一种快速测序技术,在整个基因组范围内检测和鉴定结构变异。
它包括染色质构象捕获测序(Capture Hi-C)、全基因组测序(Whole Genome Sequencing,WGS)和目标区域测序(Targeted Resequencing)。
这些方法可以对整个基因组进行全面的变异分析,从而获得准确的结构变异信息。
3. 荧光原位杂交(FISH)方法荧光原位杂交(FISH)方法是一种常用的细胞遗传学技术,可用于检测基因组结构变异。
FISH方法使用特定的探针与目标DNA序列结合,通过检测荧光信号来确定是否存在基因重排或插入变异。
这种方法对于研究染色体结构变异和染色质重排等变异是非常有价值的。
4. 单细胞测序方法单细胞测序方法可以在单个细胞水平上检测和分析基因组结构变异。
这种方法通过对单个细胞进行测序,可以获得准确的变异信息,从而揭示不同细胞之间的基因组结构差异。
单细胞测序方法在人类体细胞和肿瘤细胞中的应用已经取得了重要的突破。
下面将以两个实例来说明基因组结构变异分析的常用方法:实例一:人类基因组的结构变异研究人类基因组的结构变异对于理解人类疾病的发病机制至关重要。
ChIP-seq
Log 2normalized read count0 101212121212Replicate技术参数样品要求文库类型测序策略数据量类型分析内容项目周期 ChIP-seq45天一般深度:10~20 M clean reads; 高深度:40 M clean readsHiSeq SE50DNA文库已经富集好的DNA样品(样品总量:≥50 ng; 样品浓度:10 ng/μl)ChIP-seq 是以染色质免疫共沉淀(ChIP)为基础,基于Illumina HiSeq 2500测序平台,在全基因组范围内研究组蛋白或被转录因子结合的 DNA 区域,以高效率的测序手段得到高通量的数据结果。
参考文献[1] Ross-Innes C S, Stark R, Teschendorff A E, et al . Differential oestrogen receptor binding is associated with clinical outcome in breast cancer [J]. Nature, 2012, 481(7381): 389-393.[2] Ricardi M M, Zhong S, et al . Genome-wide data (ChIP-seq) enabled identification of cell wall-related and aquaporin genes as targets of tomato ASR1, a drought stress-responsive transcription factor [J]. BMC plant biology, 2014, 14(1): 29.案例解析[案例一] 雌激素受体结合异常与乳腺癌临床试验的相关性研究[1]雌激素受体(ER)是一种能与基因结合的物质,其受体复合物可移位到细胞核结合下游基因并激活它们转录,为肿瘤生长提供助力。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
卢卡斯 沃特曼 Lukas Wartman
华盛顿大学研究癌症的博士卢卡斯.沃特曼罹患有白血病。2003年被确诊时,
沃特曼年仅25岁,将要从圣路易斯的华盛顿大学医学院毕业。命运的齿轮突然
错开一格,沃特曼被迫离开医学院,拖着病躯挪至医院。历经两年的强化和维持
化疗后,沃特曼似乎痊愈了。但就在距当初发病的5年后,白血病突然再度发作。
研究表明,白血病患者复发,生存几率只有4%~5%。幸运的是,在接受来自
弟弟的骨髓移植后,沃特曼又顺利地度过3年。
2011年,沃特曼病情第三次发作。这一次,甚至连存活几率的统计都没有
了。医生虽施以化疗、骨髓注射等治疗方案,结果均宣告无效。沃特曼的病情迅
速恶化,没有任何已知的手段能治好他。
这时,沃特曼在圣路易斯华盛顿大学基因组研究中心的同事们出马了。他们
成功申请将沃特曼纳入一项研究项目,计划通过分析导致其患上成人急性淋巴性
白血病的基因来制订新的治疗方案。癌症由体细胞基因突变诱发,但此前,还从
未有人彻底研究过上述癌症相关基因变异的形成。通过对沃特曼的癌细胞和正常
细胞的基因组进行完全测序和比较,研究者们试图弄清楚,沃特曼的基因究竟是
哪里出了问题。
接下去的几周时间里,沃特曼的同事们全力投入这场生死营救中。华盛顿大
学里26台基因测序仪器中的一台,以及一台超级计算机,为沃特曼昼夜不停地
运行。奇迹真的发生了!研究者们找到了元凶:一个正常基因FLT3表达过于活
跃,刺激了沃特曼癌细胞的快速生长增殖。更令人欣慰的是,他们找到了一种可
能抑制问题基因表达的新药,这种新药之前只被批准用于治疗晚期肾癌。沃特曼
成了第一个用这种药治疗白血病的病人。
目前,经历九死一生的沃特曼病情已有好转。没人敢说他已经得到治愈,但
至少,沃特曼度过了去年的危险期,依旧活于人世。
糖尿病
目前常见的有一型还有二型糖尿病,但其实还有一种叫MODY的糖尿病,它不能精确
诊断,约10%1型或者2-5%2型糖尿病其实为MODY,被误诊。
而针对不同的型别,使用的治疗方式也是不同的。
类型 基因 临床特征 对应治疗
MODY 1(5%) HNF4A
随时间加重,幼儿期可能有低血糖 磺脲类,胰岛素
MODY
2(22%)
GCK
症状轻或无,持续轻微高血糖,控制饮食运动 健康的生活方式,降糖
药无效,不需要胰岛素
MODY
3(58%)
HNF1A
随时间加重,低的肾糖阈值,增加
心血管疾病发生的机率
磺脲类,胰岛素
MODY 4<1% IPF1
胰腺发育不全 磺脲类,胰岛素
MODY 5(2%) HNF1B
肾囊肿,先于糖尿病出现
胰岛素,其他的特殊治
疗
MODY 6<1% NeuroD1
胰岛素
人体对于所有药物,在多次使用之后均会产生耐药性,所以个性化针对性的合理治疗可
极大的增强治疗效果