全基因组重测序分析
基因组重测序

基因组重测序背景介绍 全基因组重测序,是对基因组序列已知的个体进行基因组测序,并在个体或群体水平上进行差异性分析的方法。
与已知序列比对,寻找单核苷酸多态性位点(SNP )、插入缺失位点(InDel ,Insertion/Deletion )、结构变异位点(SV ,Structure Variation )位点及拷贝数变化(CNV) 。
可以寻找到大量基因差异,实现遗传进化分析及重要性状候选基因的预测。
涉及临床医药研究、群体遗传学研究、关联分析、进化分析等众多应用领域。
随着测序成本的大幅度降低以及测序效率的数量级提升,全基因组重测序已经成为研究人类疾病及动植物分子育种最为快速有效的方法之一。
利用illumina Hiseq 2000平台,将不同插入片段文库和双末端测序相结合,可以高效地挖掘基因序列差异和结构变异等信息,为客户进行疾病研究、分子育种等提供准确依据。
重测序的两个条件:(1)该物种基因组序列已知;(2)所测序群体之间遗传性差异不大( >99% 相似度 )在已经完成的全基因组测序及其基因功能注释的基础上,采用全基因组鸟枪法(WGS )对DNA 插入片段进行双末端测序。
技术路线生物信息学分析送样要求1.样品总量:每次样品制备需要大于5ug 的样品。
为保证实验质量及延续性,请一次性提供至少20ug的样品。
如需多次制备样品,按照制备次数计算样品总量。
2.样品纯度:OD值260/280应在1.8~2.0 之间;无蛋白质、RNA或肉眼可见杂质污染。
3.样品浓度:不低于50 ng/μL。
4.样品质量:基因组完整、无降解,电泳结果基因组DNA主带应在λ‐Hind III digest 最大条带23 Kb以上且主带清晰,无弥散。
5.样品保存:限选择干粉、酒精、TE buffer或超纯水一种,请在样品信息单中注明。
6.样品运输:样品请置于1.5 ml管中,做好标记,使用封口膜封好;基因组DNA如果用乙醇沉淀,可以常温运输;否则建议使用干冰或冰袋运输,并选择较快的运输方式。
全基因组重测序数据分析详细说明

全基因组重测序数据分析详细说明全基因组重测序(whole genome sequencing, WGS)是一种高通量测序技术,用于获取个体的整个基因组信息。
全基因组重测序数据分析是指对这些数据进行处理、分析和解读,以获得有关个体的遗传变异、基因型、表达和功能等信息。
下面详细说明全基因组重测序数据分析的过程和方法。
首先,全基因组重测序数据的质量控制是必不可少的。
这一步骤包括对测序数据进行质量评估、剔除低质量序列,并进行去除接头序列和过滤序列等预处理操作,以确保后续分析的准确性和可靠性。
接下来,需要对全基因组重测序数据进行序列比对,将读取序列与参考基因组进行比对,以确定每个读取序列在参考基因组上的位置。
常用的比对工具包括Bowtie、BWA、BLAST等。
比对的结果将提供每个读取序列的基因组位置信息。
在序列比对完成后,就可以进行个体的变异检测。
变异检测的目的是识别个体的单核苷酸多态性(single nucleotide polymorphisms, SNPs)、插入缺失变异(insertions/deletions, indels)和结构变异(structural variations, SVs)等基因组变异。
通常,变异检测分为两个步骤:变异发现和变异筛选。
变异发现即根据比对结果,通过一定的算法和统计学原理,找到潜在的变异位点。
然后,利用临床数据库、已知变异数据库和基因功能注释数据库等,进行变异筛选,剔除假阳性和无功能变异,筛选出最有可能的致病变异。
接着,对筛选出的变异位点进行基因型確定。
基因型的确定可以通过直接从比对结果中读取碱基信息,或者通过再次测序来获取高度精确的基因型,以获得更可靠的变异信息。
随后,对变异位点进行注释和功能预测。
注释是指对变异位点进行功能和可能影响的基因、基因组区域和调控元件等进行注释。
常用的注释工具包括ANNOVAR、SnpEff、VEP等。
功能预测则是根据变异位点的位置和可能影响的功能进行预测,如是否影响蛋白质功能、是否在编码序列、是否在启动子或增强子区域等。
全基因组重测序技术的原理与进展

全基因组重测序技术的原理与进展全基因组重测序技术(Whole Genome Sequencing,WGS)是一种高通量的DNA序列分析技术,它可以检测出基因组中所有的DNA序列,包括基因及非编码区域的DNA序列,从而得到生物体的完整基因组信息。
全基因组重测序技术的应用范围极广,涵盖了医学、农业、生态、进化等领域。
全基因组重测序技术的原理是通过高通量测序技术对DNA样本进行多次、高精度的测序,将测序结果进行拼接处理,从而得到基因组的完整DNA序列。
目前常见的高通量测序技术包括Illumina、PacBio、ONT等,它们各自有优势和不足。
其中Illumina技术常用于重测序主流的生物体基因组,所需测序的覆盖度较高; PacBio和ONT均具有较长的单次读长,对于检测基因组中较长的插入或缺失变异等具有一定优势。
此外,针对富集序列的RNA测序技术也可以用于特定基因的全基因组重测序。
全基因组重测序技术的应用范围极广。
在医学领域,全基因组重测序技术被广泛应用于遗传病和肿瘤研究,可用于检测基因突变、引起复杂疾病的复杂基因组变异、疾病个体间的基因表达差异。
在农业领域,全基因组重测序技术可用于育种改良、农药研发、疫苗疾病预测和品种鉴定等。
在生态系统学与进化生物学研究中,全基因组重测序技术可用于物种间基因组比较、种群遗传学研究、进化历程研究等。
在全基因组重测序技术的基础上,个性化基因组医学逐渐发展。
通过对人类的基因组进行全基因组重测序,可以获得具体人群的基因突变情况和遗传倾向,从而进行个性化的病症预测和治疗方案设计,这在未来可能成为临床诊疗工具的一部分。
全基因组重测序技术的快速发展,也催生了大量为全基因组重测序应用领域所开发出的生物信息学工具。
生物信息学工具对于全基因组重测序技术的应用至关重要,它们可以对高通量测序数据进行高效准确地解析,分析复杂的基因组变异,对基因功能进行详细分析,从而推动基因组学领域的快速发展。
全基因组重测序数据分析

全基因组重测序数据分析1. 数据质量控制:对测序数据进行质量控制,包括去除低质量的碱基、过滤含有接头序列和接头污染的序列等。
这一步骤可以使用各种质控工具,例如FastQC、Trim Galore等。
2. 比对到参考基因组:将经过质控的测序数据与参考基因组进行比对。
参考基因组一般是已知的物种的基因组序列,在人类研究中通常使用人类参考基因组。
比对工具主要有BWA、Bowtie等。
3. 变异检测:从比对结果中检测出样本与参考基因组之间的差异,称为变异检测。
这包括单核苷酸变异(SNV)、插入/缺失(Indel)、结构变异(SV)等。
常用的变异检测工具有GATK、SAMtools、CNVnator等。
4. 注释和解读:对检测到的变异进行注释和解读,以确定其对基因功能和疾病相关性的影响。
注释可以包括基因、转录本、蛋白质功能、通路、疾病关联等信息。
常用的注释工具包括ANNOVAR、Variant Effect Predictor等。
5.结果可视化:将分析结果以图表或图形的形式展示出来,以便研究人员更好地理解和解释结果。
常用的可视化工具包括IGV、R软件等。
除了上述步骤,全基因组重测序数据分析还可以应用于其他研究领域,例如种群遗传学、复杂疾病研究、药物研发等。
在进行这些研究时,可能还需要其他分析方法和工具来完成特定的研究目标。
总之,全基因组重测序数据分析是一个复杂而关键的过程,它可以帮助研究人员了解个体的基因组特征,并揭示与疾病发生和发展相关的重要信息。
在不断发展的测序技术和分析方法的推动下,全基因组重测序数据分析将在基因组学领域中发挥越来越重要的作用。
全基因组重测序技术在疾病诊断中的应用

全基因组重测序技术在疾病诊断中的应用引言:全基因组重测序(whole-genome sequencing,WGS)是一项先进的技术,可以对个体的整个基因组进行高通量、高分辨率的测序。
随着测序技术的不断发展和成本的降低,全基因组重测序已经成为许多疾病诊断和治疗中的重要工具。
本文将探讨全基因组重测序技术在疾病诊断中的应用。
一、儿童遗传性疾病的诊断儿童遗传性疾病是指由遗传突变引起的各种罕见疾病。
由于这些疾病表现复杂多样,单一基因突变引起不同临床表型,传统方法很难准确诊断。
而全基因组重测序技术可以快速而精确地鉴定突变位点,并了解患者携带的致病变异情况。
通过对家系及相关资料进行综合分析,可以更精准地判断是否为染色体异常或单基因突变所致,从而为儿童遗传性疾病的诊断提供更准确的依据。
二、肿瘤基因组学研究全基因组重测序技术在肿瘤基因组学研究中具有重要意义。
肿瘤是由一系列DNA 突变和表观遗传异常引起的复杂疾病,因此了解患者的个体基因组信息对精准治疗至关重要。
全基因组重测序可以检测出肿瘤样本中所有突变位点,包括常见和罕见变异,在进一步分析突变驱动机制、变异负荷以及预后评估方面有着不可替代的作用。
此外,全基因组重测序技术还可以帮助发现新型靶向治疗标志物,并指导个性化治疗方案的制定。
三、个体化药物治疗随着全基因组重测序技术的应用,越来越多的医生开始使用“个体化药物治疗”来提高治愈率和降低患者副作用。
通过对患者进行基因组测序并与已经积累的大量数据库进行比对,可以预测疾病和药物反应的关联。
在使用特定药物之前,医生可以预测药物是否有效、是否会引起不良反应,并据此制定个体化的治疗方案。
这种精确的用药策略可以提高治疗效果,减少药物副作用,使患者获得更好的治疗结果。
四、遗传性疾病筛查与婚姻匹配全基因组重测序技术还可以应用于遗传性疾病筛查和婚姻匹配中。
通过对患者进行基因组测序,可以及早发现致病基因突变,并向有关人士提供相关信息以指导受孕决策。
动植物全基因组重测序简介

全基因组重测序是对已知基因组序列的物种进行不同个体的基因组测序,并在此基础上对个体或群体进行差异性分析。
基于全基因组重测序技术,人们可以快速进行资源普查筛选,寻找到大量遗传变异,实现遗传进化分析及重要性状候选基因的预测。
随着测序成本降低和拥有参考基因组序列物种增多,全基因组重测序成为动植物育种和群体进化研究迅速有效的方法。
简化基因组测序技术是对与限制性核酸内切酶识别位点相关的DNA进行高通量测序。
RAD-seq(Restriction-site Associated DNA Sequence)和GBS (Genotyping-by-Sequencing)技术是目前应用最为广泛的简化基因组技术,可大幅降低基因组的复杂度,操作简便,同时不受参考基因组的限制,可快速鉴定出高密度的SNP位点,从而实现遗传进化分析及重要性状候选基因的预测。
简化基因组技术尤其适合于大样本量的研究,可以为利用全基因组重测序技术做深度信息挖掘奠定坚实的基础。
全基因组重测序和简化基因组测序技术可广泛应用于变异检测、遗传图谱构建、功能基因挖掘、群体进化等研究,具有重大的科研和产业价值。
产品脉络图。
全基因组关联分析-基于全基因组重测序
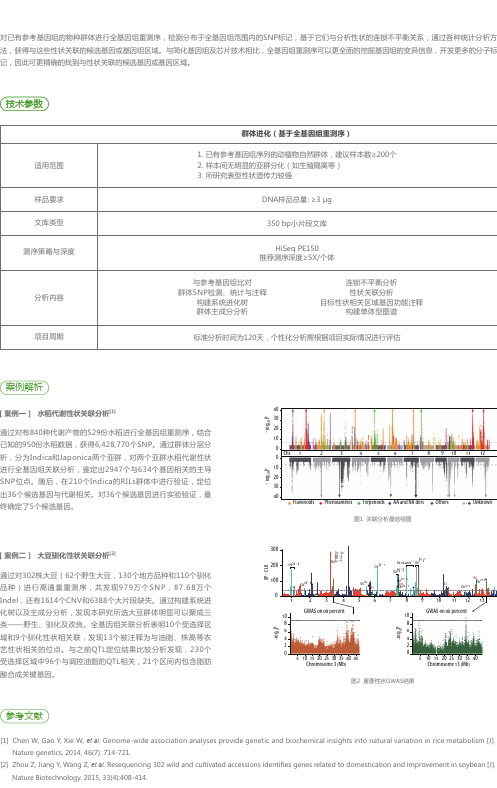
图2 重要性状GWAS结果
参考文献
[1] Chen W, Gao Y, Xie W, et al. Genome-wide association analyses provide genetic and biochemical insights into natural variation in rice metabolism [J]. Nature genetics, 2014, 46(7): 714-721.
对已有参考基因组的物种群体进行全基因组重测序,检测分布于全基因组范围内的SNP标记,基于它们与分析性状的连锁不平衡关系,通过各种统计分析方 法,获得与这些性状关联的候选基因或基因组区域。与简化基因组及芯片技术相比,全基因组重测序可以更全面的挖掘基因组的变异信息,开发更多的分子标 记,因此可更精确的找到与性状关联的候选基因或基因区域。
ቤተ መጻሕፍቲ ባይዱ
与参考基因组比对 群体SNP检测、统计与注释
构建系统进化树 群体主成分分析
连锁不平衡分析 性状关联分析
目标性状相关区域基因功能注释 构建单体型图谱
标准分析时间为120天,个性化分析需根据项目实际情况进行评估
案例解析
[案例一] 水稻代谢性状关联分析[1]
通过对有840种代谢产物的529份水稻进行全基因组重测序,结合 已知的950份水稻数据,获得6,428,770个SNP。通过群体分层分 析,分为Indica和Japonica两个亚群,对两个亚群水稻代谢性状 进行全基因组关联分析,鉴定出2947个与634个基因相关的主导 SNP位点。随后,在210个Indica的RILs群体中进行验证,定位 出36个候选基因与代谢相关。对36个候选基因进行实验验证,最 终确定了5个候选基因。
全基因组重测序数据分析

全基因组重测序数据分析全基因组重测序是一种高通量测序技术,可以获取一个个体的整个基因组的序列信息。
全基因组重测序数据分析是从这些序列数据中提取有用信息的过程,包括基因组装、变异检测和功能注释等。
本文将详细介绍全基因组重测序数据分析的步骤和一些常用的分析方法。
全基因组重测序数据分析的第一步是基因组装。
基因组装是指将测序得到的片段序列根据其重叠关系拼接成连续的序列。
目前有许多基因组装软件可供选择,如SOAPdenovo和SPAdes等。
这些软件会将测序片段根据其序列重叠情况进行集成,以获取最长的连续序列。
基因组装后,下一步是进行变异检测。
变异是指个体基因组与参考基因组之间的差异,可以分为单核苷酸变异(SNV)和结构变异(SV)两种类型。
SNV是指个体基因组中的单个碱基发生改变,包括单碱基插入、缺失和替换等。
SV则是指较大的基因组片段发生改变,包括插入、缺失、倒位和重组等。
变异检测的主要目标是通过比对个体的测序数据与参考基因组的序列,识别和注释这些变异。
为了提高变异检测的准确性,通常需要进行数据预处理和质量控制。
数据预处理包括去除接头序列、低质量序列和重复序列等,以提高后续分析的准确性和效率。
质量控制则是评估测序数据的质量,如测序深度、覆盖度和错误率等,以保证分析结果的可靠性。
除了变异检测,全基因组重测序数据还可以用于其他类型的分析,如基因表达分析和基因组结构分析。
基因表达分析可以通过比对测序数据和转录组数据库,识别并定量基因的表达水平。
基因组结构分析可以揭示染色体水平的变异和基因组结构的演化。
这些分析可以帮助研究人员研究基因组的功能和进化等问题。
总之,全基因组重测序数据分析是一个复杂的过程,涉及到多个步骤和分析方法。
通过对测序数据的组装、变异检测和功能注释等分析,可以获得有关个体基因组的详细信息,为基因功能研究和遗传疾病诊断提供重要参考。
随着测序技术的不断发展,全基因组重测序数据分析将会变得更加高效和准确。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
全基因组重测序分析主要包括样品准备和测序、数据比对、多态性分析等步骤。
1)准备样品材料和测序。
根据研究目的确定是单个个体测序,还是多个个体或群体测序,估计所需的测序深度,确定测序读长和文库大小,选择合适的测序平台。
具体所需测序深度取决于测序错误率,参考基因组组装质量,物种的连锁不平衡程度以及研究目的。
一般来说,如果主要是研究个体或群体单个位点核苷酸差异(SNP),每个样品的测序深度通常为2-6倍;如果是检测大片段的DNA序列差异一般需要20?30倍的基因组覆盖深度;如果是用于全基因组关联分析基因分型,所需的覆盖深度可以降到0.5倍CHuang et al 2010)。
2)数据读段比对。
首先检测测序数据的质量,根据质量确定是否要对数据进行质量控制和读段末端截短等比对预处理措施。
然后将数据比对到所研究物种的参考基因组上或其近缘物种的参考基因组上。
鉴于传统的基于局部比对算法的工具无法快速准确的比对短片段序列,生物信息家们发了一系列基于二代测序数据的短序列比对工具。
根据算法的不同,这些工具主要分为两类,一类基于空位种子索引法
3) DNA序列多态性分析:DNA序列多态性分析是重测序分析的基础,主要包括识别单核苷酸多态性(SNP),短片段的插入和删除(InDel),结构变异(StructuralVariation, SV, 一般定义为大于50bp的序列变异),拷贝数变异(Copy NumberVariation,CNV)等。
广义上,拷贝数变异也可以看做是一种结构变异。