Small $P_{perp}^2$ Elastic Proton-Proton Polarization near 300 GeV
_大型强子对撞机_撞开发现之门

1000亿个质子组成, 但在对撞点上, 束团的尺寸只有银针大小:长不过 几厘米, 粗细仅有16 微米 (大约相 流的速度可达光速的99.9999991%。 当于最细的头发丝)在圆环的四个 。 每个质子携带的能量将达到 7 万亿 对撞点上, 这些银针一根接一根通
29
2 0 0 8 ・9
Modern science
相遇事件就已经发生了。 位于探测 器不同层中的元件, 能够对穿过该 元件的一些特定粒子做出特有的响 应。每个事例都将产生大约 1 兆字 节(M B )的数据,两秒钟就是 1 皮字节(P B ,相当于 1 0 亿 M B ) , 这些数据流将通过上百万条通讯信 道传送出来。 拥有多个级别的触发系统会把 洪水般的数据减少到可以控制的程 度。 初级触发将收集和分析探测器 中部分子系统的数据, 根据一些独 立要素挑选出其中最有价值的事例。 比如, 如果一个高能μ子的飞行径 迹与束流轴的夹角偏大, 这个事例 就会被选中。这个所谓的“初级触 发” 由嵌合在硬件中的数百个专用
器将记录并测量每次对撞产生出的 上千个粒子。 尽管这些探测器尺寸 巨大, 安装精度却要求极高, 一些部 件必须定位在50微米的精度以内。 在两个最大的探测器中, 每一 个都拥有近 1 亿条数据流,每秒钟 产生的数据能够写满10万张光盘— —只需要 6 个月的时间,这些光盘 就可以从地球堆到月球。因此,这
科技长廊
KE JI CHANG LANG
过,每秒钟发生 6 亿多次粒子对
些实验不会去记录所有的数据, 而
展开。此前,一个扇区曾被冷却、 加电,然后又回到室温。这些扇区 先接受单独检测, 然后再进行整体 试运行。一旦通过检验,一个质子 束流就会注入圆环, 沿着两条束流 管中的一条, 在周长27千米的圆环 中运转。 向LHC主环提供束流的一系列 小型加速器已通过验收, 它们可以 把能量为0.45万亿电子伏特的质子 注入 LHC。束流的第一次注入将是 关键一步,LHC 的科学家们将先注 入低强度束流,以降低 LHC 硬件损 毁的风险。只有对测试束流在 LHC 内部的运行情况进行仔细评估, 并 对引导磁场进行精确修正之后, 科 学家们才会注入强度更高的束流。 LHC首次在7万亿电子伏特的设计能 量下运行时, 在圆环中顺 时针和逆时针方向绕行的 质子束团都只有一个。 最 终满负荷运转时, 每个方 向上将有约 3000 个质子 束团绕行。 如此慎重地对LHC加 速器展开全面调试, 肯定 会查出许多问题。 工程师 和科学家们需要多少时间 来解决这些问题, 目前还 无法预知。 但如果一个扇 区必须回到室温下进行维 修,LHC 的启动时间又将 推迟好几个月。 LHC 上的 4 个巨型探 测器各负责一项实验, 分 别被称为 A T L A S 、 ALICE、CMS 和 LHCb。它
600kV ns 脉冲中子发生器的一些相关问题的研究

第31卷第4期原子能科学技术V o l.31,N o.4 1997年7月A tom ic Energy Science and T echno logy Ju ly1997600kV n s脉冲中子发生器的一些相关问题的研究张立山 关遐令 毛孝勇 沈冠仁(中国原子能科学研究院核物理研究所,北京,102413)就中子物理飞行时间技术对脉冲中子发生器的需要,对中国原子能科学研究院600kV n s脉冲中子发生器的聚束和切割性能的某些参量进行分析和计算,将结果与实验进行比较,并把它们用于脉冲化装置的调试。
关键词 脉冲 聚束器 切割器脉冲化中子源是利用飞行时间方法进行核物理实验的重要装置之一,尤其在中子物理的快中子微分和双微分截面的测量中更是必不可少。
它克服了伴随粒子法测量时间长和双闪烁体法对散射样品的限制,但它对脉冲束的束流品质提出了相当高的要求。
在现有条件下为获得高精度的实验数据,对本脉冲化装置的要求是:脉冲半宽在112n s以内、脉冲底宽和半宽比为3—5、重复频率在115M H z左右,束流平均强度约为30—50ΛA。
为确保束流品质稳定地达到以上标准,对脉冲化装置的聚束器和切割器的主要技术参量进行计算,以便得到聚束器的最佳聚束电压值、相位和幅度的稳定度,同时确定最佳切割宽度和切割引起能散与离子源初始发射度的关系,推导脉冲半宽与离子源初始能散的对应关系,这对实验调试和运行是有重要意义的。
1 单漂移聚束器的参数计算111 聚束的基本原理一束有一定能量差别的粒子,要求它们经过一段距离后能同时到达靶上,则它们的能量E t和相应起始时刻t的关系为:E t=E0 (1-t Σ)2(1)式中,E0为中心粒子的初始能量,Σ为中心粒子从零时刻起到靶上所需时间。
一般在tνΣ时有如下关系:∃E=E t-E0≈E0(1+2t Σ-1)=2tE0 Σ(2)∃E称为调制能量。
由式(2)可知∃E与t近似成线性关系。
由t、t′(t′=d t d L)相空间中相点运动方程可知:收稿日期:1996211229 收到修改稿日期:1997201217d t ′t ′=-F Z (t ′)2m d z =-d E 2E(3)t ′1t ′0=(E 0E 1)12(4)在Υ、Υ′(Υ′=d Υ d L )相空间中,令∆0=∃E E 0,中心粒子Υc =0,Υ′c =0;对任意粒子有:Υ0=Ξ(t 0-t c )=Ξt 0(5)Υ′0=Ξ(t ′0-t ′c )≈Ξt ′c ∆0 2(6)在粒子受正弦波电压调制时,粒子能量为:E =E 0+V m sin Υ0(7)其中:V m 为调制电压幅度。
反应堆物理题库
西安交通大学——核反应堆物理分析(共470题)从反应堆物理的角度看,良好的慢化剂材料应具有什么样的性能?答案:慢化剂是快中子与它的核发生碰撞后能减速成热中子的材料,这与它的三种中子物理性能有关:δ-平均对数能量缩减;Σs-宏观散射截面;Σa-宏观吸收截面。
综合评价应是δ和Σs都比较大而Σa又较小的材料才是较好的慢化材料,定量地用慢化能力δΣs和慢化比δ和Σs/Σa来比较。
试列出常用慢化剂的慢化能力和慢化比。
核力所具有的特点是什么?答案:基本特点是:核力是短程力,作用范围大约是1~2×10-13cm;核力是吸引力,中子与中子,质子与中子,质子与质子之间均是强吸引力。
核力与电荷无关。
核力具有饱和性,每一核子只与其邻近的数目有限的几个核子发生相互作用。
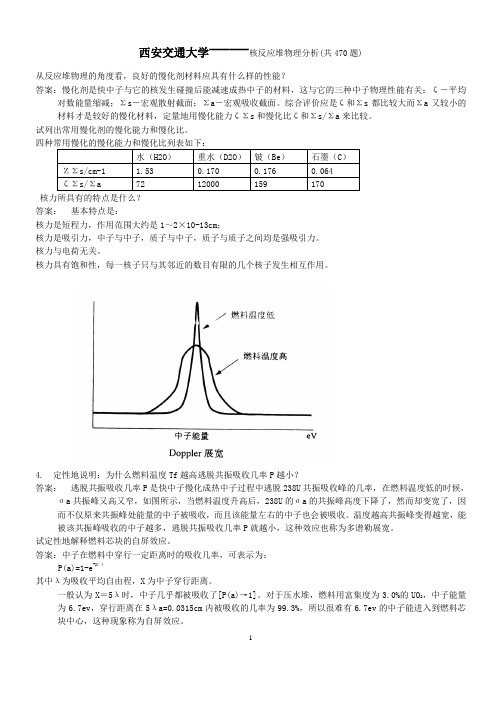
4. 定性地说明:为什么燃料温度Tf越高逃脱共振吸收几率P越小?答案:逃脱共振吸收几率P是快中子慢化成热中子过程中逃脱238U共振吸收峰的几率,在燃料温度低的时候,ζa共振峰又高又窄,如图所示,当燃料温度升高后,238U的ζa的共振峰高度下降了,然而却变宽了,因而不仅原来共振峰处能量的中子被吸收,而且该能量左右的中子也会被吸收。
温度越高共振峰变得越宽,能被该共振峰吸收的中子越多,逃脱共振吸收几率P就越小,这种效应也称为多谱勒展宽。
试定性地解释燃料芯块的自屏效应。
答案:中子在燃料中穿行一定距离时的吸收几率,可表示为:P(a)=1-e-X/λ其中λ为吸收平均自由程,X为中子穿行距离。
一般认为X=5λ时,中子几乎都被吸收了[P(a)→1]。
对于压水堆,燃料用富集度为3.0%的UO2,中子能量为6.7ev,穿行距离在5λa=0.0315cm内被吸收的几率为99.3%,所以很难有6.7ev的中子能进入到燃料芯块中心,这种现象称为自屏效应。
6. 什么是过渡周期?什么是渐近周期?答案:在零功率时,当阶跃输入-正反应性ρ0(ρ0<β)后,反应堆功率的上升速率(或周期)是随ρ0输入后的时间t而改变的(如图所示)。
质子实验

质子百科名片质子(proton)是一种带 1.6 × 10-19 库仑(C)正电荷的亚原子粒子,直径约 1.6 to 1.7×10?15 m 1,质量是938百万电子伏特/c?(MeV/c?),即1.6726231 × 10-27 kg,大约是电子质量的1836.5倍。
质子属于重子类,由两个上夸克和一个下夸克通过胶子在强相互作用下构成。
原子核中质子数目决定其化学性质和它属于何种化学元素。
目录科学含义1. 稳态2. 历史3. 应用4. 反质子5. 负质子6. 基本信息7. 基本性质8. 质子各国的读法9. 关于中子态的形成10. 质子的发现11. 质子理论12. 质子守恒文学含义1. 解释2. 历史典籍中的记录科学含义1. 稳态2. 历史3. 应用4. 反质子5. 负质子6. 基本信息7. 基本性质8. 质子各国的读法9. 关于中子态的形成10. 质子的发现11. 质子理论12. 质子守恒文学含义1. 解释2. 历史典籍中的记录展开编辑本段科学含义质子(proton)是一种带 1.6 × 10-19 库仑(C)正电荷的亚原子粒子,直径约 1.6 to 1.7×10?15 m [1],质量是938百万电子伏特/c²(MeV/c²),即1.6726231 × 10-27kg,大约是电子质量的1836.5倍。
质子属于重子类,由两个上夸克和一个下夸克通过胶子在强相互作用下构成。
原子核中质子数目决定其化学性质和它属于何种化学元素。
氢原子最常见的同位素1H 的原子核由一个质子构成。
其它原子的原子核则由质子和中子在强相互作用下构成。
稳态至今为止质子被认为是一种稳定的、不衰变的粒子。
但也有理论认为质子可能衰变,只不过其寿命非常长。
到今天为止物理学家没有能够获得任何可能理解为质子衰变的实验数据。
水中的氢离子绝大多数都是水合质子。
《CALPUFF模式用于放射性核素不同尺度的迁移扩散研究》

《CALPUFF模式用于放射性核素不同尺度的迁移扩散研究》篇一一、引言随着核能利用的普及,放射性核素迁移扩散的问题越来越受到关注。
在放射性核素的研究中,其不同尺度的迁移扩散是一个关键的科学问题。
近年来,计算流体动力学模型如CALPUFF在放射性核素迁移扩散的研究中得到了广泛的应用。
本文将详细探讨CALPUFF模式在放射性核素不同尺度的迁移扩散研究中的应用。
二、CALPUFF模式简介CALPUFF是一种基于高斯烟羽模型的气象模型,适用于模拟大气中污染物的长距离传输和扩散。
它利用大气环境中的物理、化学和气象条件来模拟放射性核素的迁移和扩散过程,并可根据不同尺度的研究需求调整模型参数。
三、CALPUFF模式在放射性核素迁移扩散研究中的应用1. 大尺度迁移扩散研究在大尺度迁移扩散研究中,CALPUFF模式能够有效地模拟放射性核素在大气中的长距离传输和扩散过程。
通过调整模型参数,可以更好地反映不同气象条件对放射性核素迁移扩散的影响。
此外,CALPUFF模式还可以与其他地理信息系统(GIS)相结合,实现空间数据的可视化处理和分析。
2. 中尺度迁移扩散研究在中尺度迁移扩散研究中,CALPUFF模式能够更精细地模拟放射性核素在局部地区的扩散过程。
通过建立详细的地理和环境信息数据库,可以更准确地模拟放射性核素在不同地形、气象条件下的迁移和扩散过程。
这对于评估局部地区放射性核素污染风险和制定相应的防护措施具有重要意义。
3. 小尺度迁移扩散研究在小尺度迁移扩散研究中,CALPUFF模式可以用于模拟放射性核素在微观尺度上的迁移和扩散过程。
这有助于研究放射性核素在土壤、水体等介质中的迁移和转化过程,为评估核事故等紧急情况下的放射性核素污染风险提供科学依据。
四、案例分析以某核事故为例,我们利用CALPUFF模式对该事故中放射性核素的迁移和扩散过程进行了模拟。
通过调整模型参数,我们成功地模拟了放射性核素在不同时间、不同空间尺度的迁移和扩散过程。
串联亲和层析 protocol

UNIT19.20 Strep/FLAG Tandem Affinity Purification(SF-TAP)to Study Protein InteractionsChristian Johannes Gloeckner,1Karsten Boldt,1,2and Marius Ueffing1,21Helmholtz Zentrum M¨u nchen,Neuherberg,Germany2Technical University of Munich,Munich,GermanyABSTRACTIn recent years,several methods have been developed to analyze protein-protein interac-tions under native conditions.One of them,tandem affinity purification(TAP),combinestwo affinity-purification steps to allow isolation of high-purity protein complexes.Thisunit presents a methodological workflow based on an SF-TAP tag comprising a doubletStrep-tag II and a FLAG moiety optimized for rapid as well as efficient tandem affinitypurification of native proteins and protein complexes in higher eukaryotic cells.Depend-ing on the stringency of purification conditions,SF-TAP allows both the isolation ofa single tagged-fusion protein of interest and purification of protein complexes undernative conditions.Curr.Protoc.Protein Sci.57:19.20.1-19.20.19.C 2009by John Wiley&Sons,Inc.Keywords:SF-TAP r tandem affinity purification r protein complexesINTRODUCTIONThe analysis of protein-protein interactions under native conditions has been a challengeever since immunoprecipitation(IP)became a common methodology.Low yields andnonspecific binding of proteins have been associated with IP.On the other hand,IPfacilitates targeted analysis of protein interactions with respect to a predefined proteinof interest,given that a suitable antibody is available that features monospecificity andselectivity for this protein.Tandem affinity purification(TAP;UNIT19.19)can significantly reduce the backgroundcaused by nonspecific binding of proteins,as it combines two affinity purifications basedon two different affinity matrices(Rigaut et al.,1999).TAP has been widely used topurify protein complexes from different species(Collins and Choudhary,2008).The TAPtechnique was originally developed to analyze the yeast protein interactome(Gavin et al.,2002).Although the original TAP tag,consisting of a Protein A-tag,a TEV(tobacco etchvirus)protease cleavage site,and a calmodulin binding peptide(CBP)tag,has alreadybeen successfully used in mammalian cells(Bouwmeester et al.,2004),several featuresof thisfirst-generation tag remain suboptimal,such as its high molecular mass(21kDa),the dependency on proteolytic cleavage,and CBP,which may interfere with calciumsignaling within eukaryotic cells.This unit presents an alternative TAP protocol for theisolation of protein complexes from higher eukaryotic cells.The Strep/FLAG tandemaffinity purification(SF-TAP)tag(Gloeckner et al.,2007)combines a tandem Strep-tagII(Skerra and Schmidt,2000;Junttila et al.,2005)and a FLAG tag,resulting in a small4.6-kDa tag.Both moieties have a medium affinity and avidity to their immobilizedbinding partners.Therefore,the tagged fusion proteins and their binding partners canbe recovered under native conditions without the need for time-consuming proteolyticcleavage.In thefirst step,desthiobiotin is used for elution of the SF-TAP fusion proteinfrom the Strep-Tactin matrix.In the second step,the FLAG octapeptide is used for elutionof the SF-TAP fusion protein from the anti-FLAG M2affinity matrix.An overview of the Current Protocols in Protein Science19.20.1-19.20.19,August2009Published online August2009in Wiley Interscience().DOI:10.1002/0471140864.ps1920s57Copyright C 2009John Wiley&Sons,Inc.Identification of Protein Interactions19.20.1 Supplement57Strep/FLAGTandem AffinityPurification (SF-TAP)19.20.2Supplement 57Current Protocols in Protein Science A B 1. purification 2. purification binding to Strep-Tactin binding to FLAG matrix elution with desthiobiotin elution with FLAG peptide Key:SF-TAP desthiobiotin FLAG peptide Figure 19.20.1The S trep/FLAG ta n dem affin ity p u rificatio n .(A )N-a n d C-termi n al S F-T AP ta gs (POI,protei n of i n tere s t).(B )Overview of both p u rificatio n s tep s .(1)P u rificatio n by the ta n dem S trep-ta g II moiety:bi n di ng to S trep-T acti n matrix followed by el u tio n with de s thiobioti n .(2)P u rificatio n by the FLAG-ta g moiety:bi n di ng to a n ti-FLAG M2affin ity matrix followed by el u -tio n with FLAG peptide.Abbreviatio ns :s p.,s pecific i n teractor s (s how n a s g ray circle s );n .s p.,n o ns pecific protei ns (co n tami n a n t s ;s how n a s white circle s ).SF-TAP technique and the tag sequence is shown in Figure 19.20.1.The SF-TAP protocol represents an efficient,fast and straightforward purification of protein complexes from mammalian cells within 2hr.This unit describes the full workflow,starting with the cell culture work needed for recombinant expression of the SF-TAP fusion proteins,followed by the SF-TAP protocol (see Basic Protocol 1)and ending with mass spectrometric analysis of the samples (see Basic Protocol 4).Special focus is given to the crucial step of sample preparation for mass spectrometry.For the identification of associated proteins following SF-TAP,the volume of the SF-TAP eluates is reduced by ultrafiltration using centrifugal units with a low molecular weight cut-off or by chloroform/methanol precipitation (see Support Protocol 2).The samples are then directly subjected to proteolytic digestion (see Basic Protocol 2)for analysis on a nano liquid chromatography (LC)–coupled electron sprayIdentification of Protein Interactions 19.20.3Current Protocols in Protein Science Supplement 57Figure 19.20.2Flow chart of a S F-T AP approach i n cl u di ng M S ide n tificatio n of cop u rified pro-tei ns .Thi s figu re co nn ect s all protocol s pre s e n ted i n thi s un it.tandem mass spectrometer.For complex samples,which contain many proteins,an alternative procedure for SDS-PAGE pre-fractionation is provided,including a method for sensitive MS-compatible Coomassie protein staining (see Support Protocol 3)followed by in-gel proteolytic digestion (see Basic Protocol 3).By reducing sample complexity,pre-fractionation helps to increase the number of protein identifications on state-of-the-art LC-coupled tandem mass spectrometers.Representative MS-analysis protocols are provided for an Orbitrap mass spectrometer (Thermo Fisher Scientific),a fast and sensitive system allowing high identification rates from SF-TAP purifications even with low amounts of protein in the sample (see Basic Protocol 4).Finally,a strategy for meta analysis of mass spectrometric data sets using the Scaffold software is provided (see Support Protocol 4).It can generally be used for the analysis of large MS/MS data sets.Figure 19.20.2provides a flowchart of the entire analytical process.Strep/FLAGTandem AffinityPurification (SF-TAP)19.20.4Supplement 57Current Protocols in Protein ScienceBASICPROTOCOL 1STREP/FLAG TANDEM AFFINITY PURIFICATION (SF-TAP)OF PROTEIN COMPLEXES FROM HEK293CELLS A flowchart of the SF-TAP procedure is shown in Figure 19.20.3.Materials HEK293cells (ATCC no.CRL-1573)Complete DMEM containing 10%FBS (APPENDIX 3C )SF-TAP vectors with appropriate insert,and empty control plasmid (see Critical Parameters)Negative control (see annotation to step 3,below)Transfection reagent of choice (see UNIT 5.10)Phosphate-buffered saline (PBS;APPENDIX 2E ),prewarmed Lysis buffer (see recipe)Strep-Tactin Superflow resin (IBA GmbH,cat.no.2-1206-10)Tris-buffered saline (TBS;see recipe)Wash buffer (see recipe)Desthiobiotin elution buffer:dilute 10×buffer E (IBA GmbH,cat.no.2-1000-025)1:10in H 2O (final concentration,2mM desthiobiotin)Anti–FLAG M2agarose (Sigma-Aldrich)FLAG elution buffer (see recipe)14-cm tissue culture plates Cell scraper Millex GP 0.22-μm syringe-driven filter units (Millipore)End-over-end rotator Microspin columns (GE Healthcare,cat.no.27-3565-01)End-over-end rotator Microcon YM-3centrifugal filter devices (Millipore)Additional reagents and equipment for transfection of mammalian cells (UNIT 5.10)Transfect HEK293cells 1.Seed HEK293cells on 14-cm plates at ∼1–2×107cells per dish in complete DMEM medium containing 10%FBS.The amount of cells used for SF-TAP purification can be varied depending on the ex-pression levels of the bait ually,four 14-cm dishes,corresponding to a final amount of ∼4×108HEK293cells,is a good starting point.Strong overexpression of the bait protein usually increases copurification of heat-shock proteins such as HSP70.For in-depth analysis,it is therefore recommended to generate cell lines stably expressing the bait protein.See Support Protocol 1for a stable transfection method.2.Grow cells overnight.3.Transfect cells with the SF-TAP plasmids using a transfection reagent of choice (according to manufacturer’s protocols).HEK293cells can be easily transfected with lipophilic transfection reagents.The trans-fection efficiency is usually >80%.For a typical SF-TAP experiment,1to 4μg plasmid per 14-cm dish is used.Depending on the cell type other transfection reagents may be favorable (also see UNIT 5.10).Although SF-TAP purifications typically exhibit low background caused by nonspecific binding of proteins to the affinity matrix,a suitable negative control should be used in every experiment.Cells transfected with the empty expression vectors may be used in the same amount as for the SF-TAP-tagged bait protein.However,the tag is quite small and expressed at low levels if not fused to a protein.Thus,the untransfected cell line is an acceptable,simple,and inexpensive alternative for a negative control.Identification of Protein Interactions 19.20.5Current Protocols in Protein Science Supplement 571-4 × 108 HEK293 cell s(1-4 co n fl u e n t 14-cm plate s )expre ss i ng S F-TAP f us io n protei nly s i s(15 mi n 4C)vol u mered u ctio nce n trif ug atio n (10 mi n 10,000 × g )a n aly s i sretai n su per n ata n t fi n alel u atei n c u batio n with50 μl/plate S trep-Tacti n matrix (1 hr)el u tio n with200 μl FLAGel u tio n b u ffer(10 mi n )wa s h 3 time s with 500 μl wa s h b u ffer (s pi n 5 s ec, 100 × g )wa s h 3 time s with500 μl wa s h b u ffer(s pi n 5 s ec, 100 × g )el u tio n with 500 μl de s thiobioti n el u tio n b u ffer (10 mi n )i n c u batio n with25 μl/platea n ti-FLAG M2a g aro s e(1 hr)Figure 19.20.3Flow chart for the S F-T AP proced u re.4.Let cells grow for 48hr.If necessary,cells can be starved in DMEM without FBS for 12hr prior to harvesting.Starving might be desirable if cell signaling is to be analyzed,especially prior to differ-ential treatment with growth factors,to eliminate effects of serum growth factors.Lyse cells5.Remove medium from the plates.6.Optional:Rinse cells in warm PBS.Strep/FLAGTandem AffinityPurification (SF-TAP)19.20.6Supplement 57Current Protocols in Protein Science7.Scrape off cells in 1ml lysis buffer per 14-cm plate on ice using a cell scraper,and combine lysates from each experimental condition in a 1.5-ml microcentrifuge tube.8.Lyse cells by incubating 15min on ice with mixing by hand from time to time.9.Pellet cell debris,including nuclei,by centrifuging 10min at 10,000×g ,4◦C.10.Clear lysate supernatant by filtration through a 0.22-μm syringe filter.Perform SF-TAP 11.Wash Strep-Tactin Superflow resin twice,each time with 4resin volumes TBS and once with 4resin volumes lysis buffer.12.Incubate lysates with 50μl per 14-cm plate of settled Strep-Tactin Superflow resin for 1hr at 4◦C (use an end-over-end rotator to keep the resin evenly distributed).Note that a maximum of 200μl settled resin per spin column should not be exceeded.If more than four 14-cm plates (∼4×108HEK293cells)are used,reduce the volume per plate or use additional spin columns in step 13.13.Centrifuge for 30sec at 7000×g ,4◦C,remove the supernatant until 500μl remains,and transfer resin to a microspin column.Snap off bottom closure of the spin column prior to use.The maximum volume of the spin columns is 650μl.Alternatively,centrifugations for wash and elution steps can be performed at room temperature if no cooled centrifuge is available.14.Remove remaining supernatant by centrifugation in the spin column for 5sec at 100×g ,then wash resin three times,each time with 500μl wash buffer (centrifuge 5sec at 100×g each time to remove the supernatant)at 4◦C.Replug spin columns with inverted bottom closure prior to adding the elution buffer in step 15.IMPORTANT NOTE:Do not allow the resin to run dry.Depending on the bait protein,this markedly reduces the yield.15.Add 500μl desthiobiotin elution buffer and gently mix the resin by hand for 10min on ice.16.Remove the plug of the spin column,transfer the column to a new collection tube,and collect the eluate by centrifuging 10sec at 2000×g ,4◦C.If spin columns were closed by the top screw cap during incubation with elution buffer,the cap needs to be removed prior to centrifugation,to allow the pressure to balance out.17.Wash anti–FLAG M2agarose resin three times,each time with 4resin volumes TBS.Suspend resin in TBS and transfer it to microspin columns,then remove the buffer by centrifuging 5sec at 100×g .25μl settled resin per 14-cm plate will be needed.18.Transfer eluate from step 16corresponding to each 14-cm plate to a microspin column containing 25μl settled anti-FLAG M2agarose prepared as in step 17.19.Plug columns,close columns with top screw caps,and incubate for 1hr at 4◦C (on an end-over-end rotator).20.Wash once with 500μl wash buffer,and then twice,each time with 500μl TBS (centrifuge 5sec at 100×g each time to remove the supernatant)at 4◦C.21.For elution,incubate with 4bead volumes (at least 200μl)FLAG elution buffer for 10min,keeping the columns plugged and gently mixing the resin several times.22.After incubation,remove the plugs and top screws of the spin columns,transfer to new collection tubes,and collect the eluate(s)by centrifugation (10sec at 2000×g ).Identification of Protein Interactions 19.20.7Current Protocols in Protein Science Supplement 5723.Depending on downstream method to be used,either precipitate protein (see SupportProtocol 2)or concentrate the eluate by Microcon YM-3centrifugal filter units according to manufacturer’s protocols.SUPPORT PROTOCOL 1GENERATION OF HEK293CLONES STABLY EXPRESSINGSF-TAP-TAGGED PROTEINSIn Basic Protocol 1,SF-TAP-tagged proteins are transiently expressed.However,strong overexpression of the bait protein usually increases copurification of heat-shock proteins such as HSP70.For in-depth analysis,it is therefore recommended to generate cell lines stably expressing the bait protein.This protocol presents a quick method for generating stable HEK293lines.MaterialsHEK293cells (ATCC no.CRL-1573)Complete DMEM containing 10%FBS (APPENDIX 3C )SF-TAP vectors with appropriate insert,and empty control plasmid (see Critical Parameters)Transfection reagent of choice (see UNIT 5.10)Phosphate-buffered saline (PBS;APPENDIX 2E )Complete DMEM medium (APPENDIX 3C )G418(PAA Laboratories, )Freezing solution:90%fetal bovine serum (FBS;Invitrogen)/10%dimethylsulfoxide (DMSO;AR grade)Lysis buffer (see recipe)Blocking reagent:5%(w/v)nonfat dry milk in TBS (see recipe for TBS)containing 0.1%(v/v)Tween 20Anti-FLAG M2antibody (Sigma-Aldrich)10-cm tissue culture dishes12-well and 6-welll tissue culture platesCentrifuge2-ml cryovials (Nunc)Additional reagents and equipment for transfection of mammalian cells (UNIT 5.10),trypsinization and counting of cells (UNIT 5.10),and immunoblotting (UNIT 10.10)Grow and transfect cells1.Grow cells in complete DMEM containing 10%FBS.2.Transfect cells with expression plasmid using a transfection reagent of choice ac-cording to the manufacturer’s protocols.3.Change medium after 6hr.Select cells4.After 48hr,trypsinize and count cells (APPENDIX 3C )and seed them at low density (1×106cells per 10-cm dish)to allow formation of single colonies upon selection.5.Add G418(500to 1000μg/ml)for selection of the SF-TAP expression vectors,which are based on pcDNA3.0and contain a neomycin-resistance gene.6.Grow the cells under G-418selection for 2to 4weeks,changing the medium every second day.7.Collect single colonies with a 200-μl pipet into 12-well plates.8.Keep colonies under G418selection until the cell density is sufficient for expanding them to 6-well dishes (two wells per clone).Strep/FLAGTandem AffinityPurification (SF-TAP)19.20.8Supplement 57Current Protocols in Protein ScienceCryopreserve cells 9.Grow cells to >90%confluency and trypsinize (APPENDIX 3C )one well of each clone for generation of cryostocks.10.Generate cryostocks:a.Wash cells from one well once by adding 3ml PBS,centrifuging 5min at 800×g ,room temperature,and resuspending the pellet in 500μl freezing buffer.b.Transfer resuspended cells to 2-ml cryovials.c.Freeze cells slowly:keep cells for 1hr at −20◦C,then overnight at −80◦C,followed by storage in a liquid nitrogen tank.For cultivation and expansion of confirmed clones,thaw the cryostock at 37◦C,wash cells once with medium,and plate cells onto 10-cm culture dishes.Test for expression of bait protein 11.Lyse one well of each clone in 300μl lysis buffer and test for expression of the bait protein by immunoblotting (UNIT 10.10).SF-TAP proteins can be detected using the anti-FLAG M2antibody (Sigma-Aldrich)at a dilution of 1:1000to 1:5000in blocking reagent.SUPPORTPROTOCOL 2CHLOROFORM/METHANOL PRECIPITATION OF PROTEINS The chloroform/methanol precipitation method described by Wessel and Fl¨u gge (1984)precipitates proteins with high efficiency and yields samples containing low levels of salt contamination.Materials SF-TAP eluate (from Basic Protocol 1)Methanol (AR grade)Chloroform (AR grade)2-ml polypropylene sample tubes 1.Transfer 200μl SF-TAP eluate to a 2-ml sample tube.All steps are performed at ambient temperature.2.Add 0.8ml of methanol,vortex,and centrifuge for 20sec at 9000×g ,room temperature.3.Add 0.2ml chloroform,vortex,and centrifuge for 20sec at 9000×g ,room temperature.4.Add 0.6ml of deionized water,vortex for 5sec,and centrifuge for 1min at 9000×g ,room temperature.5.Carefully remove and discard the upper layer (aqueous phase).The protein precipitate (visible as white flocks)is in the interphase.6.Add 0.6ml of methanol,vortex,and centrifuge for 2min at 16,000×g ,room temperature.7.Carefully remove the supernatant and air dry the pellet.The pellet can be stored for several months at –80◦C.Identification of Protein Interactions 19.20.9Current Protocols in Protein Science Supplement 57BASIC PROTOCOL 2IN-SOLUTION DIGEST OF PROTEINS FOR MASS SPECTROMETRIC ANALYSISThe in-solution digest described here is a quick and efficient method to digest the SF-TAP eluate after protein precipitation (Support Protocol 2).The use of an MS-compatible surfactant helps to solubilize the precipitated proteins.In order to allow the identification of cysteine-containing peptides,random oxidation is prevented,rather than reverted,by applying a DTT/iodoacetamide treatment prior to digestion,leading to a defined-mass adduct.The digested protein sample can then be directly subjected to analysis on an LC-coupled tandem mass spectrometer.MaterialsPrecipitated protein (see Support Protocol 2)50mM ammonium bicarbonate (freshly prepared)RapiGest SF (Waters):prepare 2%(10×)stock solution in deionized water 100mM DTT (prepare from 500mM stock solution;store stock up to 6months at −20◦C)300mM iodoacetamide (prepare fresh)50×(0.5μg/μl)trypsin stock solution (Promega;store at −20◦C)Concentrated (37%)HCl60◦C incubatorPolypropylene inserts (Supelco,cat.no.24722)1to 200μl gel-loader pipet tips (Sorenson Bioscience,/contact.cfm )1.Dissolve the protein pellet in 30μl of 50mM ammonium bicarbonate by extensive vortexing.2.Add 3μl of 10×(2%)RapiGest stock solution (final concentration,0.2%).RapiGest (sodium 3-[(2-methyl-2-undecyl-1,3-dioxolan-4-yl)methoxyl]-1-propanesulfo-nate)is an acid-labile surfactant that helps to solubilize and denature proteins to make them accessible to proteolytic digestion (Yu et al.,2003).3.Add 1μl of 100mM DTT and vortex.4.Incubate 10min at 60◦C.5.Cool the samples to room temperature.6.Add 1μl of 300mM iodoacetamide and vortex.7.Incubate for 30min at room temperature.Samples should be protected from light,since iodoacetamide is light-sensitive.8.Add 2μl trypsin stock solution and vortex.9.Incubate at 37◦C overnight.10.Add 2μl of concentrated (37%)HCl to hydrolyze the RapiGest.For hydrolysis of the RapiGest reagent,the pH must be <2.11.Transfer samples to polypropylene inserts (remove spring).12.Incubate for 30min at room temperature.13.Place inserts in 1.5-ml microcentrifuge tubes and microcentrifuge 10min at 13,000×g ,room temperature.One hydrolysis product of the RapiGest reagent is water-immiscible and can be removed by centrifugation.After centrifugation,it is visible as faint film (oleic phase)on top of theStrep/FLAGTandem Affinity Purification (SF-TAP)19.20.10Supplement 57Current Protocols in Protein Science aqueous sample phase.The other hydrolysis product is an ionic water-soluble component which does not interfere with reversed phase LC or MS analysis.A white pellet might appear.14.Carefully recover the solution between the upper oleic phase and the pellet using gel-loader tips.The sample can now be directly subjected to C18HPLC separation prior to MS/MS-analysis (LC-MS/MS;Basic Protocol 4).Pre-fractionation (Basic Protocol 3)is optional.BASIC PROTOCOL 3PRE-FRACTIONATION VIA SDS-PAGE AND IN-GEL DIGESTION PRIOR TO LC-MS/MS ANALYSIS Pre-fractionation prior to MS analysis increases the number of peptides which can be an-alyzed,and therefore the peptide coverage of identified proteins.This benefit is achieved by overcoming the undersampling problem mainly caused by the limited capacity of the trapping columns used in nano–LC chromatography,or that occurs with high complexity.For these samples,SDS-PAGE pre-fractionation can be used to reduce the complexity.For less complex samples or samples with low protein content,the in-solution digest (Basic Protocol 2)is preferred.Materials Protein sample (e.g.,from Basic Protocol 1or Support Protocol 2)10%NuPAGE gels (Invitrogen)MOPS running buffer (Invitrogen)40%and 100%acetonitrile (AR grade;prepare fresh)5mM DTT (prepare from 500mM stock;store stock up to 6months at −20◦C)25mM iodoacetamide (prepare fresh)Digestion solution:dilute 50×trypsin stock solution (0.5μg/μl,Promega)1:50in 50mM ammonium bicarbonate (freshly prepared)1%and 0.5%(v/v)trifluoroacetic acid (TFA;prepare fresh from 10%v/v stock)50%(v/v)acetonitrile/0.5%(v/v)TFA (prepare fresh)99.5%(v/v)acetonitrile/0.5%(v/v)TFA (prepare fresh)2%(v/v)acetonitrile/0.5%(v/v)TFA Concentration units (e.g.,Microcon from Millipore)Scalpel Polypropylene 96-well microtiter plate:polystyrene material should be avoided since,depending on the product,polymers can be extracted from plastics which produce strong background signals in mass spectrometry 60◦C incubator or heating block Polypropylene 0.5-ml reaction tubes Microtiter plate shaker (e.g.,V ortex mixer equipped with microtiter-plate adaptor)HPLC sample tubes Additional reagents and equipment for SDS-PAGE (UNIT 10.1)and colloidal Coomassie blue staining of gels (Support Protocol 3)Prepare samples 1.Concentrate samples using concentration units (e.g.,Microcon).2.Supplement samples with Laemmli loading buffer (SDS-PAGE loading buffer;UNIT 10.1).A detailed description of the SDS gel electrophoresis and standard buffers can be found in UNIT 10.1or in the protocols supplied with the NuPAGE system.Identification of ProteinInteractions19.20.11Perform electrophoresis and stain gels3.Separate samples on 10%NuPAGE gels according to the manufacturer’s protocols,using MOPS running buffer.4.Stop electrophoresis after the gel front has travelled 1to 2cm.5.Stain gels with colloidal Coomassie blue (see Support Protocol 3).Avoid strong staining of the bands since it increases the time necessary for destaining.6.Excise desired gel pieces with a clean scalpel (three to ten slices,depending on the complexity of the sample).Destain and process gel slices7.Transfer gel pieces into individual wells of a 96-well plate.8.Wash by adding 100μl water to each well and incubating for 30min.9.For destaining:a.Wash twice,each time by incubating the gel slices for 10min in 100μl/well of 40%acetonitrile.b.Wash for 5min in 100μl/well of 100%acetonitrile (if gels are still blue,repeat de-staining).10.Add 100μl of 5mM DTT,then incubate 15min at 60◦C in an incubator or heatingblock.11.Remove DTT solution and cool the plate to room temperature.12.Add 100μl per well of freshly prepared 25mM iodoacetamide,then incubate 30minin the dark.13.Wash twice,each time for 10min with 100μl/well of 40%acetonitrile.14.Wash 5min with 100μl/well of 100%acetonitrile.15.Discard supernatant and air dry (or SpeedVac)the gel pieces to complete dryness.Digest and extract gel slices16.Add 20to 30μl per well of freshly prepared digestion solution (depending on the sizeof the gel plugs).Wrap plates in Parafilm to reduce evaporation during the overnight incubation (or use a humidified incubator in step 17).17.Digest overnight at 37◦C.18.For extraction of the peptides from the gel piece,add 10μl 1%TFA,then shake15min on a V ortex mixer with a microtiter plate adapter.The peptides are extracted in three steps with increasing acetonitrile concentrations (steps 18to 23).19.Transfer liquid (extract 1)to a 0.5-ml polypropylene tube.20.Add 50μl 50%acetonitrile/0.5%TFA to the gel piece and shake 15min on a V ortexmixer with a microtiter plate adapter.21.Remove the liquid (extract 2)and pool extracts 1and 2.22.Add 50μl 99.5%acetonitrile/0.5%TFA to the gel piece,then shake 15min on aV ortex mixer with a microtiter plate adapter.23.Remove the liquid (extract 3)and pool extract 3with 1and 2.Strep/FLAG Tandem AffinityPurification(SF-TAP)19.20.1224.Dry samples to complete dryness in a SpeedVac evaporator.25.Redissolve samples in50μl of2%acetonitrile/0.5%TFA by shaking(e.g.,on aV ortex mixer)for10to15min,then transfer the sample into HPLC sample tubes for LC-MS/MS analysis.SUPPORT PROTOCOL3QUICK MS-COMPATIBLE COLLOIDAL COOMASSIE STAIN OF PROTEINS AFTER SDS-PAGE SEPARATIONThe colloidal Coomassie stain(Kang et al.,2002)represents a fast and sensitive MS-compatible protein staining method.In contrast to the classical staining protocol,no intense and time-consuming destaining is needed to visualize protein bands.Therefore, this method is ideal for a quick staining of the protein bands and provides good orientation on how the gel can be fractionated without splitting predominant bands(see Basic Protocol3).MaterialsElectrophoresed SDS gel containing protein samples of interest(e.g.,from Basic Protocol3)Colloidal Coomassie staining solution(see recipe)Destaining solution:10%(v/v)ethanol/2%(v/v)orthophosphoric acidGel staining trays of appropriate size1.Wash gels twice,each time for10min in deionized water in a staining tray.The SDS must be removed before staining to reduce background signals.2.Incubate gels for10min in colloidal Coomassie staining solution.The incubation steps are kept short for the staining of gels used for pre-fractionation.The staining can be prolonged up to overnight.The maximum staining will be reached after ∼3hr incubation in the staining solution.3.Incubate gels for10min in destaining solution.4.Wash gels twice,each time for10min in deionized water.BASIC PROTOCOL4LC-MS/MS ANALYSIS OF DIGESTED SF-TAP SAMPLESThe following protocol describes MS analysis of digested protein samples on an LC-coupled ESI tandem mass spectrometer.The representative MS-analysis protocol is provided for an Orbitrap mass spectrometer(Thermo Fisher Scientific).The Orbitrap system combines fast data acquisition with high mass accuracy and is therefore ideal for the analysis of SF-TAP samples.Background information on mass spectrometric analysis can be found in UNIT16.11.MaterialsDigested protein sample,either from in-solution digest(Basic Protocol2)or in-gel digest(Basic Protocol3)Nano HPLC loading buffer:0.1%formic acid in HPLC-grade waterNano HPLC buffer A:2%acetonitrile/0.1%formic acid in HPLC-grade waterNano HPLC buffer B:80%acetonitrile/0.1%formic acid in HPLC-grade water HPLC vials(Dionex)Nano HPLC system(UltiMate3000,Dionex)equipped with a trap column (100μm i.d.×2cm,packed with Acclaim PepMap100C18resin,5μm,100◦A;Dionex)and an analytical column(75μm i.d.×15cm,packed with AcclaimPepMap100C18resin,3μm,100◦A;Dionex)Mass spectrometer:Oritrap XL with a nanospray ion source(ThermoFisher Scientific;also see UNIT16.11)。
基本物理常数表
PrefaceFundamental Physical Constants: 1998Peter J. Mohr and Barry N. TaylorNational Institute of standards and Technology, Gaithersburg, MD 20899-8401This table gives the 1998 self-consistent set of values of the basic constants and conversion factors of physics and chemistry recommended by the Committee on Data for Science and Technology (CODATA) for international use. Further, it describes in detail the adjustment of the values of the subset of constants on which the complete 1998 set of recommended values is based. The 1998 set replaces its immediate predecessor recommended by CODATA in 1986. The new adjustment, which takes into account all of the data available through 31 December 1998, is a significant advance over its 1986 counterpart. The 1998 adjustment was carried out by P. J. Mohr and B. N. Taylor of the National Institute of Standards and Technology (NIST) under the auspices of the CODATA Task Group on Fundamental Constants. The standard uncertainties (i.e., estimated standard deviations) of the new recommended values are in most cases about 1/5 to 1/12 and in some cases 1/160 times the standard uncertainties of the corresponding 1986 values. Moreover, in almost all cases the absolute values of the differences between the 1998 values and the corresponding 1986 values are less than twice the standard uncertainties of the 1986 values.The Task Group was established in 1969 with the aim of periodically providing the scientific and technological communities with a self-consistent set of internationally recommended values of the fundamental physical constants based on all applicable information available at a given point in time. The first set was published in 1973 and was followed by a revised set first published in 1986; the current 1998 set first appeared in 1999. In the future, the CODATA Task Group plans to take advantage of the high level of automation developed for the current set in order to issue a new set of recommended values at least every four years.Relative std. Quantity Symbol Value Unit uncert.u ra Value recommended by the Particle Data Group,Caso et al.,Eur.Phys.J.C3(1-4),1-794(1998).b Based on the ratio of the masses of the W and Z bosons mW/m Z recommended by the Particle Data Group(Caso et al.,1998).The value for sin2θW they recommend,which is based on a particular variant of the modified minimal subtraction(。
Geant4 Tutorial Course 2009
Geant4 Tutorial Course 2009
G4NDL (Geant4 Neutron Data Library)
• • • The neutron data files for High Precision Neutron models The data are including both cross sections and final states. The data are derived evaluations based on the following evaluated data libraries (in alphabetic order)
CrossSection [barn]
1 G4 ENDF
0.1
0.01
0.001
Elastic
Inelastic
Inelastic
Inelastic
Inelastic
Inelastic
(n,nγ)
(n,2n)
(n,nα)
(n,np)
(n,p)
(n,α)
Geant4 Tutorial Course 2009
– – ENDF/B-VI.8
• • • • • 313 isotopes including 5 isomers 15 elements Released on 2006 Dec almost 400 isotopes not yet migrated
– –
How to write Nuclear Data files How to use the Nuclear Data files
– – – – isotropic emission discrete two-body kinematics N-body phase-space distribution continuum energy-angle distributions
原子物理学杨福家1-6章_课后习题答案
原子物理学课后前六章答案(第四版)杨福家著(高等教育出版社)第一章:原子的位形:卢瑟福模型第二章:原子的量子态:波尔模型第三章:量子力学导论第四章:原子的精细结构:电子的自旋第五章:多电子原子:泡利原理第六章:X射线第一章习题1、2解1.1 速度为v的非相对论的α粒子与一静止的自由电子相碰撞,试证明:α粒子的最大偏离角约为10-4rad.要点分析: 碰撞应考虑入射粒子和电子方向改变.并不是像教材中的入射粒子与靶核的碰撞(靶核不动).注意这里电子要动.证明:设α粒子的质量为Mα,碰撞前速度为V,沿X方向入射;碰撞后,速度为V',沿θ方向散射。
电子质量用me表示,碰撞前静止在坐标原点O处,碰撞后以速度v沿φ方向反冲。
α粒子-电子系统在此过程中能量与动量均应守恒,有:(1)ϕθααcos cos v m V M V M e +'= (2)ϕθαsin sin 0v m V M e -'= (3)作运算:(2)×sin θ±(3)×cos θ,(4)(5)再将(4)、(5)二式与(1)式联立,消去V’与v,化简上式,得(6)θϕμϕθμ222s i n s i n )(s i n +=+ (7)视θ为φ的函数θ(φ),对(7)式求θ的极值,有若 sinθ=0, 则θ=0(极小)(8)(2)若cos(θ+2φ)=0 ,则θ=90º-2φ(9)将(9)式代入(7)式,有θϕμϕμ222)(90si nsi nsi n+=-θ≈10-4弧度(极大)此题得证。
1.2(1)动能为5.00MeV的α粒子被金核以90°散射时,它的瞄准距离(碰撞参数)为多大?(2)如果金箔厚1.0 μm,则入射α粒子束以大于90°散射(称为背散射)的粒子数是全部入射粒子的百分之几?要点分析:第二问是90°~180°范围的积分.关键要知道n, 注意推导出n值.其他值从书中参考列表中找.解:(1)依金的原子序数Z2=79答:散射角为90º所对所对应的瞄准距离为22.8fm.(2)解: 第二问解的要点是注意将大于90°的散射全部积分出来.(问题不知道nA,但可从密度与原子量关系找出)从书后物质密度表和原子量表中查出ZAu=79,AAu=197, ρAu=1.888×104kg/m3依θa2 sin即单位体积内的粒子数为密度除以摩尔质量数乘以阿伏加德罗常数。
p300诱导的乙酰化修饰参与脂多糖诱导的炎症介质合成
p 300诱导的乙酰化修饰参与脂多糖诱导的炎症介质合成胡柯1,曹湘玉1,李玉娴1,刘灵丽1,陈月富1,陈立军1,黄民江1,谭碧峰2,尹辉明3湖南医药学院1医学院,2第一附属医院心血管内科,3第一附属医院呼吸与危重症医学科,湖南怀化418000Coactivator p300-induced H3K27acetylation mediates lipopolysaccharide-induced inflammatory mediator synthesisHU Ke 1,CAO Xiangyu 1,LI Yuxian 1,LIU Lingli 1,CHEN Yuefu 1,CHEN Lijun 1,HUANG Minjiang 1,TAN Bifeng 2,YIN Huiming 31Medical College of Hunan University of Medicine,2Department of Cardiology,First Affiliated Hospital,Hunan University of Medicine,3Department of Respiratory and Critical Care Medicine,First Affiliated Hospital,Hunan University of Medicine,Huaihua 418000,China摘要:目的探讨辅助激活因子p300诱导的乙酰化修饰介入脂多糖(LPS )诱导的炎症介质合成过程及其作用机制。
方法Agilent Sureprint G3Mouse Gene Expression V2微阵列芯片以及蛋白免疫印迹(WB )技术联合于小鼠巨噬细胞(RAW246.7)中筛选表达水平与LPS 刺激强度相关的分子;凝胶电泳迁移实验(EMSA )以及染色质免疫共沉淀(chip-qpcr )方法验证相关分子与炎症基因IL-6以及TNF-α启动子部位的结合现象;相关分子过表达或干扰质粒转染RAW246.7后,WB 法检测转染质粒的作用效果,ELISA 方法检测IL-6以及TNF-α合成水平,染色质免疫沉淀-测序技术(chip-seq )分析炎症基因启动子部位相关分子的结合以及H3K27的乙酰化修饰水平。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
a
r
X
i
v
:
h
e
p
-
p
h
/
9
6
1
0
5
3
7
v
1
3
0
O
c
t
1
9
9
6
hep-ph/9610537
SmallP2⊥ElasticProton-ProtonPolarizationnear300GeV
A.D.Krisch1andS.M.Troshin
2
chiralsymmetryisspontaneouslybroken.InReggetheorythePomeronisusually
assumedtobehelicityconserving;howeverthisassumptionhasnotbeentested.
Moreoveritwasshownthatunitarity,whichistheabsorptivecorrection,generates
differentphasesinthehelicity-flipandnonflipPomeroncontributions[7].TheCNI
analysingpowerwouldcertainlychangeiftherewereanysignificanthadronicsingle-
helicity-flipamplitude.Thus,wewilltrytousetheexistingexperimentaldataon
theanalysingpowerinproton-protonelasticscatteringtoestimatethehadronic
single-helicity-flipamplitudenearElab=300GeV,whereonemightuseaCNIpo-
larimeterwithapolarizedprotonbeam[8,9].
Instandardcalculations,theCNIanalyzingpowerarisesthroughtheinterference
betweentheCoulombhelicity-single-flipamplitudefC5andthehadronic(Nuclear)
helicity-nonflipamplitudesfN1andfN3.However,therecouldalsobeanon-zero
hadronichelicity-single-flipamplitudefN5,whichcanbeparameterizedby
τ(s,t)≡
m
−t
|fN5|
−t.Moreover,atsmallt,theimaginarypartoff
N
2
ispropor-
tionalto∆σt,whichisthedifferencebetweenthetransverse-spintotalcross-sections,
σtot(↑↑)−σtot(↑↓);thisdifferenceisprobablysmallathighenergies[10].
WeshallalsoassumethatfN1=fN3atsmallt,sincetheexisting12GeVdata
onAllissmall,andtheexisting∆σldataissmallanddecreaseswithenergy.Thus,
forpurelyhadronicprocessesatsmallt,
Andσs(s−4m2)Im[fN1fN∗5],(3)
where
dσ
s(s−4m2)
|fN1|2.(4)
Notethat,athighenergyandsmall−t,thespin-nonflipamplitudefN1isprimar-
ilyimaginarybecausetheelastichadronicnonflipscatteringisprimarilythediffrac-
tiveshadowscatteringduetothedominantinelasticprocesses;thus,fN1≃i|fN1|.
However,thespin-flipamplitudefN5canhavebotharealandanimaginarypart;
writingitasfN5=|fN5|eiφ5,wehave
Andσs(s−4m2)|fN1||fN5|cosφ5,(5)
whereφ5isthephase.Notethatφ5=π/2correspondstoapureimaginaryhelicity-
single-flipamplitudewhilethephases0andπcorrespondtoapurerealfN5.
2
NowbycombiningEqs.(4)and(5),weobtain
An=−2
|fN5|
2
√
−tneart=0.Moreover,theexperimentaldatanear
−t=0.2(GeV/c)2implyans−1energydependenceforAn;therefore,wehave
An≃A√s.(8)
TheconstantA≃3.6GeVcanbeobtainedfromtheAn=1.6/sfittotheexperi-
mentaldata.Thentheestimatefortheparameterτis
τcosφ5≃−
Am
s
.(9)
At300GeVwethenobtainτcosφ5≃0.003,whichweassumedcanbeextrapolated
from−t=0.2to0.003(GeV/c)2.
Nowwecanestimatethehadroniccontributiontothefullsinglehelicity-flip
amplitudef5=fC5+fN5,where
fC5≃−αsgF1(t)F2(t)−t.(10)
whereα=1/137andg=1.79istheproton’sanomalousmagneticmoment,while
F1(t)andF2(t)aretheproton’sformfactors.Weusethestandardexponential
parameterizationforthehadronicamplitudeathighenergiesandsmallt[2,3,5]
fN5=τ(cosφ5+isinφ5)s√8πm(11)
At300GeV,theratiooftherealtotheimaginaryforwardscatteringamplitude
squaredρ2isexperimentallywellbelow0.003[11],whileσtotisabout40mb.More-
over,theebt/2termandbothformfactorsareallverycloseto1at−t=0.003(GeV/c)2.
Thus,at300GeVand−t=0.003(GeV/c)2weobtain
f
N
5
τ(cosφ5+isinφ5)=0.003(1+i),(13)
thuswehave
|fN5|