PCR引物流程设计详解教学内容
实验五 PCR引物设计及评价

实验五 PCR引物设计及评价【实验目的】1、掌握引物设计的基本要求,并熟悉使用Primer premier5.0软件进行引物搜索。
2、掌握使用软件oligo6.0对设计的引物进行评价分析。
【实验原理】一、引物设计原则聚合梅链式反应(polymerase chain reaction)即PCR技术,是一种在体外快速扩增特定基因或DNA 序列的方法,故又称基因的体外扩增法。
PCR技术已成为分子生物学研究中使用最多,最广泛的手段之一,而引物设计是PCR技术中至关重要的一环,使用不合适的PCR引物容易导致实验失败:表现为扩增出目的带之外的多条带(如形成引物二聚体带),不出带或出带很弱,等等。
现在PCR 引物设计大都通过计算机软件进行,可以直接提交模板序列到特定网页,得到设计好的引物,也可以在本地计算机上运行引物设计专业软件。
引物设计原则如下:1、引物应在序列的保守区域设计并具有特异性。
引物序列应位于基因组DNA的高度保守区,且与非扩增区无同源序列。
这样可以减少引物与基因组的非特异结合,提高反应的特异性;2、引物的长度一般为15-30 bp。
常用的是18-27 bp,但不应大于38,因为过长会导致其延伸温度大于74℃,不适于Taq DNA聚合酶进行反应;3、引物不应形成二级结构。
引物二聚体及发夹结构的能值过高(超过4.5kcal/mol)易导致产生引物二聚体带,并且降低引物有效浓度而使PCR反应不能正常进行;4、引物序列的GC含量一般为40-60%。
过高或过低都不利于引发反应。
上下游引物的GC含量不能相差太大;5、引物所对应模板位置序列的Tm值在72℃左右可使复性条件最佳。
Tm值的计算有多种方法,如按公式Tm=4(G+C)+2(A+T);6、引物5'端序列对PCR影响不太大,因此常用来引进修饰位点或标记物。
可根据下一步实验中要插入PCR产物的载体的相应序列而确定。
7、引物3’端不可修饰。
引物3'端的末位碱基对Taq酶的DNA合成效率有较大的影响。
PCR技术的操作步骤高中生物

PCR技术的操作步骤高中生物摘要聚合酶链反应(PCR)是一种重要的分子生物学技术,广泛应用于基因工程、医学诊断和犯罪调查等领域。
本文将介绍PCR技术的操作步骤,包括材料准备、PCR反应体系的制备、PCR循环程序的设定和PCR产物分析等内容。
引言PCR技术是在体外模拟DNA的复制过程,通过放大目标DNA段,产生大量的特定DNA序列。
PCR技术的操作步骤简便、快速,并且具有高灵敏度和高特异性,因此被广泛应用于生物学研究和生物工程实践中。
PCR技术的操作步骤1. 材料准备•DNA模板:PCR的起始物质,可以是基因组DNA或DNA片段。
•引物:引物是一对寡聚核苷酸链(通常是20-30个碱基的寡核苷酸),用于在PCR 反应中选择性地扩增目标DNA。
•酶:聚合酶(如Taq聚合酶)和反式转录酶(如M-MLV逆转录酶),用于DNA扩增和反转录。
•反应缓冲液:提供适宜的pH和离子浓度,稳定PCR反应体系。
2. PCR反应体系的制备PCR反应体系由DNA模板、引物、酶和反应缓冲液组成。
制备PCR反应体系的步骤如下:•将PCR反应管标记编号,避免混淆。
•根据所需PCR反应的样品数量,按照实验设计准备相应的反应管。
•计算PCR反应物的体积,通常为20-100 μl。
•按照比例将DNA模板、引物、酶和反应缓冲液加入反应管中。
•轻轻混匀反应液,避免形成气泡。
3. PCR循环程序的设定PCR循环程序是PCR反应的核心部分,它包括一系列温度变化的步骤,以实现DNA 的分离、引物的结合和DNA的扩增。
常见的PCR循环程序大致分为三个阶段:变性、退火和扩增。
•变性阶段:将PCR反应体系加热至95℃,使DNA变性成两股单链。
•退火阶段:将温度降至50-65℃,引物与DNA模板结合,形成DNA双链。
•扩增阶段:将温度升至72℃,聚合酶活化,开始DNA扩增反应。
这个PCR循环程序通常重复20-40次,以产生大量目标DNA。
4. PCR产物分析PCR反应后,可以通过凝胶电泳和DNA测序等方法来分析PCR产物。
《PCR引物设计》课件

04
pcr引物的应用与案例分 析
pcr引物在基因克隆中的应用
01
pcr引物用于基因克隆的目的是为了获得目的基因的序列信息, 进而进行后续的基因功能和表达研究。
02
设计特异性引物,通过pcr技术,从基因或基因组中筛选出目的基因。
引物设计需考虑基因序列的特异性、扩增效率和避免非特异性
03
扩增等因素。
引物特异性优化
避免引物间的互补
引物之间不应存在互补序列,以避免形成引物二聚体或发夹 结构。
避免引物与模板扩增 和产物。
引物扩增效率的优化
引物与模板的匹配度
引物的3'端应与模板完全匹配,以提 高引物的扩增效率。
引物之间的匹配度
两个引物之间应有良好的匹配度,以 保证PCR反应的顺利进行。
引导合成
引物作为合成子链的起点,通过与 DNA聚合酶的结合,引导合成与 模板互补的DNA链。
决定产物长度
引物的设计决定了PCR产物的长度 ,通过选择合适的引物,可以控制 产物的大小和特异性。
pcr引物设计的基本原则
特异性
长度和序列
引物应与模板DNA具有高度的特异性,避 免与其他非目标DNA序列发生非特异性结 合。
pcr引物的未来发展方向与挑战
引物设计的自动化
随着生物信息学的发展,未来引物设计 可能更加自动化,减少人工干预和误差
。
标准化和质量控制
建立引物设计的标准化流程,加强引 物设计的质量控制,确保实验结果的
可靠性和可重复性。
新型引物设计策略
针对特定需求,开发新型引物设计策 略,提高PCR反应的特异性和灵敏度 。
引物灵敏度测试
03
测试引物在不同模板浓度下的扩增效率,选择灵敏度较高的引
PCR教材教学课件

数字PCR技术诞生,提高了检测灵 敏度和特异性。
2010年至今
随着测序技术的发展,高通量测序 与PCR技术结合,推动了基因组学 和表观遗传学的研究。
PCR技术的未来发展方向
01
简化操作流程
通过改进试剂和仪器,降低PCR 技术的操作难度,提高实验效率
。
03
拓展应用领域
将PCR技术应用于临床诊断、生 物安全检测、环境监测等领域, 提高检测的广泛性和实用性。
仪器选择
选择性能稳定、高效的PCR仪器, 确保实验结果的可靠性和重复性。
样品处理
优化样品处理方法,减少样品纯化 时间,提高实验效率。
04
PCR实验结果分析
结果判断
阴性结果
阳性结果
如果PCR产物条带与阴性对照一致,说明未 检测到目标基因,可能为基因未表达或表 达量极低。
如果PCR产物条带与阳性对照一致,说明成 功扩增了目标基因,基因表达量较高。
。
05
PCR技术的发展与展望
PCR技术的发展历程
1983年
Kary Mullis在Cetus公司工作时,发 明了聚合酶链式反应(PCR)技术。
1985年
PCR技术获得美国专利,并获得诺贝 尔化学奖。
1989年
PCR技术被正式命名,并得到广泛应 用。
1993年
TaqMan荧光定量PCR技术问世,实 现了PCR的定量检测。
PCR的应用
基因克隆和基因组测序
用于获取目的基因或测定基因组序列。
古生物学和考古学
用于分析古代生物遗骸的DNA。
遗传疾病的诊断
检测基因突变或异常表达。
法医学鉴定
用于个体识别和亲子鉴定等。
02
(完整word版)PCR引物流程设计详解
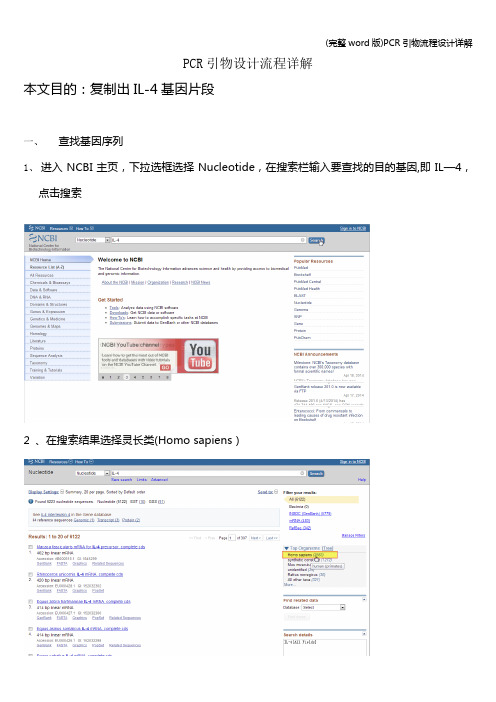
PCR引物设计流程详解本文目的:复制出IL-4基因片段一、查找基因序列1、进入NCBI主页,下拉选框选择Nucleotide,在搜索栏输入要查找的目的基因,即IL—4,点击搜索2 、在搜索结果选择灵长类(Homo sapiens)2、在灵长类IL-4基因中选择需要的mRNA序列3、查看基因的相关信息外显子区域CDs区域4、点击FASTA格式,并将序列保存到文档二、使用primer premier 5。
0设计引物1、建立新文件,将所得的序列复制进输入框内2、点击搜索按钮,搜索引物3、设置引物设计参数(因为在之前查找基因序列的时候获知,外显子区域分别为:1—200、201-248、249-425、426-618,又知在引物设计时引物位置最好跨越一个内含子,PCR产物长度通常为100—150bp,故设定上游引物位置为201—248,下游引物位置为249—425,产物长度为100-150bp)4、确认条件后,显示搜索结果4、双击选中得分最高的引物查看引物情况(上图为上游引物情况,下图为下游引物情况)5、将设计的上下游引物复制出来,保存到文档中三、使用oligo 6.0对设计的引物进行评价1、建立新文件,将从cnki上获得的cDNA复制进输入框,并点击accept接收2、接收后显示出该序列的相关信息3、点击edit按钮录入用primer设计的上游引物,每一次输入新数据后都需要点击accept按钮接收4、同理,录入下游引物5、分析上下游引物二聚体形成情况6、分析上下游引物发卡形成情况7、分析上下游引物GC%8、检测上下游引物与PCR模板其它位置错配情况9、分析PCR整体情况四、引物特异性检验(primer blast)1、进入NCBI主页,并选择blast2、选择primer blast3、在输入框内输入模板序列和上下游引物,并设定对比数据库,点击get primer进行对比4、查看blast结果Blast 结果显示,尽管IL—4与其它基因有相似区,但是引物的3’端没有完全互补。
(完整版)PCR技术(包含引物设计)

聚合酶链式反应(PCR)原理:DNA的半保留复制时,双链DNA在多种酶的作用下可以变性解链成单链,在DNA聚合酶与启动子的参与下,根据碱基互补配对原则复制成同样的两分子挎贝。
在实验条件下,DNA在高温时也可以发生变性解链,当温度降低后又可以复性成为双链。
因此,通过温度变化控制DNA的变性和复性,并设计引物做启动子,加入DNA聚合酶、dNTP就可以完成特定基因的体外复制。
PCR类似于DNA的天然复制过程,其特异性依赖于与靶序列两端互补的寡核苷酸引物。
PCR由变性 - 退火(复性)- 延伸三个基本反应步骤构成:①模板DNA的变性:模板DNA经加热至94℃左右一定时间后,使模板DNA双链或经PCR扩增形成的双链DNA解离,使之成为单链,以便它与引物结合,为下轮反应作准备;②模板DNA与引物的退火(复性):模板DNA经加热变性成单链后,温度降至40~60℃左右,引物与模板DNA单链的互补序列配对结合;③引物的延伸:DNA模板 - 引物结合物在DNA聚合酶的作用下,于72℃左右,以dNTP为反应原料,靶序列为模板,按碱基配对与半保留复制原理,合成一条新的与模板DNA链互补的半保留复制链,重复循环就可获得更多的“半保留复制链”,而且这种新链又可成为下次循环的模板。
每完成一个循环需2~4分钟,2~3小时就能将待扩目的基因扩增放大几百万倍。
PCR技术分类(常用)(1)反向PCR技术(Inverse PCR, IPCR):反向PCR是克隆已知序列旁侧序列的一种方法.主要原理是用一种在已知序列中无切点的限制性内切酶消化基因组DNA.后酶切片段自身环化.以环化的DNA作为模板,用一对与已知序列两端特异性结合的引物,扩增夹在中间的未知序列。
该扩增产物是线性的DNA片段,大小取决于上述限制性内切酶在已知基闲侧翼DNA 序列内部的酶切位点分布情况。
用不同的限制性内切酶消化,可以得到大小不同的模板DNA,再通过反向PCR获得未知片段。
PCR引物设计
上机操作7
【实验名称】 PCR引物设计 【实验目的】 掌握引物设计的基本要求,掌握使用软件 Oligo6.0进行引物设计,掌握使用软件 Bioxm2.6 进行酶切位点分析。 【实验器材】 Oligo 6.0软件,Bioxm2.6软件,NCBI 数据库
• 实验原理
PCR引物设计的目的是为了找到一对合适的核苷酸 片段,使其能有效地扩增模板DNA序列。引物设计 总体上包含三个程序:序列下载,引物设计筛选, 特异性分析。
PCR引物的设计原则:
① 引物应用核酸系列保守区内设计并具有特异性。 ② 产物不能形成二级结构。 ③ 引物长度一般在15~30碱基之间。 ④ G+C含量在40%~60%之间。 ⑤ 碱基要随机分布。 ⑥ 引物自身和互相之间不能有连续4个碱基的互补。 ⑦ 引物5′端可以修饰,引物3′端不可修饰。 ⑧ 引物 3’端的碱基要求严格配对
DraI (1260) DraI (1274) Nco I (1322)
EGFP
XhoI (2054) Sac I (2061) Not I (2331) H indIII (2063) Eco RI (2070)
MCS
Pst I (2079) Sal I (2080) K pnI (2090) Bam H I (2101) Xba I (2113)
实验步骤
1、从NCBI数据库中下载水稻CDPK1基因的序列(2238bp), 2、向Oligo6.0程序导入模板序列:复制序列后在Oligo软件的序 列展示窗口粘贴,oligo会自动去除非碱基字符。当序列输入或 粘贴完成后,点击Accept/Discard菜单中的Accept浮动命令,即 可进入引物设计模式。 3、开始引物设计:在基因的之间设计一对扩增产物长度在 1500-1800bp左右的引物,两头加上合适的酶切位点便于连入表 达载体(见后一张PPT的图)。 具体步骤自己整理并在报告上写清楚。
3_引物设计及测序结果分析(2016教案)
28对引物
搜索结果
每对引物的信息
引物分值 100分为满分
双击选中一对引物
回到主窗口
引物信息
引物及产物信息
是否出现 hairpin,dimer,false priming and cross dimer
引物编辑
引物分析
• 首先检查引物二聚体尤其是3’端二聚体 形成的可能性。 • 二项检查是发夹结构(hairpin);与 二聚体相同,发夹结构的能值越低越 好。 • 第三项检查为GC 含量,以45-55%为 宜。 • 第四False priming 检查
• 发夹结构(引物自身连续互补碱基不能大于3bp ) • 发夹结构:DNA通过自身回折使得互补的碱基对相遇,也
就是引物有部分可以自身配对
– 尤其是要避免引物3’端形成发夹结构,否则将严重影响 DNA聚合酶的延伸。 – 两引物之间不应该存在互补性,尤应避免3′端的互补重 叠以防引物二聚体的形成。一般情况下,一对引物间
8.自由能分布
• ΔG值(自由能)反映了引物与模板结合的强弱 程度。一般3’端ΔG值不要超过9(ΔG值为负 值,这里取绝对值),如此则有利于正确引发 反应而可防止错误引发。3′末端双链的ΔG是 0~-2 kcal/mol时,PCR产量几乎达到百分 之百,随着其绝对值的增加产量逐渐下降,在 -6时只有40%、到-8时少于20%、而-10 时接近于0。 • 引物的3’端的∆G 值过高,容易在错配位点形 成双链结构并引发DNA 聚合反应。
7.引物的内部稳定性
• 过去认为,引物3’端应牢牢结合在模板上才能有 效地进行延伸,故3’端最好为G或C。 • 现在的观点认为,引物的5’端应是相对稳定结构, 而3’端在碱基配对的情况下最好为低稳定性结构, 即3’端尽可能选用A或T(有说不适宜用A),少 用G或C。 • 若模板不很清楚,引物3’端最后一个碱基最好为 T,其次是G或C,而不选A,国外资料表明,当 末位为T时,即使在错配的情况下也能引发链的 合成,而末位为A时,错配时引发大大降低,G或 C居中,可见,模板很清楚时选A可以提高特异性
qPCR实验操作流程10660教学内容
qPCR实验操作流程10660教学内容q P C R实验操作流程10660Q-PCR实验流程⼀、①实验前准备,每天早上到实验室后,先把超净⼯作台的紫外灯打开15-20分钟。
②超净台前做实验,需佩戴⼲净的橡胶⼿套/⼀次性薄膜⼿套,RNA抽提需带⼝罩。
③取EP管/枪头时需⽤镊⼦,不可以⽤使⽤过的⼿套直接取⽤。
取完EP管/枪头后,袋⼦及时封好。
④橡胶⼿套须放⼊超净台照射紫外,实验操作过程中不得带出超净台,移液器在⼀天⼯作结束后调⾄最⼤量程,并⽤75%⼄醇清洁移液器,枪头盒及超净台⾯。
⑤实验进⾏的过程中或观看实验时,没有带⼝罩不要在超净台前讲话。
⼆、总RNA抽提1)细胞培养⽫中细胞样品⽤1*PBS洗两次后,⽤1ml枪将PBS吸⼲净,加⼊1ml Trizol (Invitrogen)溶液,吹打混匀,并吸⾄1.5ml RNase free EP管中使细胞充分裂解,室温静置5min;组织样品⽤液氮充分研磨,加⼊1ml Trizol (Invitrogen)溶液,混匀,室温放置5min使其充分裂解;(管盖与管壁都需标记样品名称)2)加⼊200µl氯仿,剧烈振荡混匀30s,使⽔相和有机相充分接触,室温静置3-5min;(离⼼时离⼼管按顺序排放,离⼼完毕,离⼼管的顺序也按顺序排好,与第⼀步的顺序⼀致)3) 4℃下,14,000g离⼼15min,可见分为三层,RNA在上层⽔相,移⾄另⼀个新的RNase free EP管;(⽤20-200ul的枪吸取上清,吸上清时,枪头应沿着液⾯上层吸取上清,枪头不可碰到、吸到中间层)4)沉淀RNA:加⼊等体积异丙醇,轻柔地充分混匀(颠倒6-8次)(不应⽤振荡器混匀),室温静置10min;5)4℃下,14,000g离⼼10min,收集RNA沉淀(如离⼼后仍不见EP管底部有沉淀,应将EP管放置在-80度冰箱过夜,继续在4℃下,14,000g离⼼10min,收集RNA沉淀),去上清;6)⽤75%⼄醇洗涤两次(12,000g离⼼5min)(加⼊⼄醇后只需轻轻颠倒EP管即可,不⽤振荡器震荡或枪头吸打沉淀),超净台风⼲;沉淀不能过⼲或过湿,过⼲则不易溶解,过湿则⼄醇残留。
PCR引物设计方法
PCR引物设计方法引言PCR(聚合酶链式反应)是一种常用的分子生物学技术,它能够在体外扩增DNA序列,从而使之得到大量复制。
而PCR引物是PCR反应中起到引导DNA扩增的关键组分。
本文将介绍PCR引物的设计方法,帮助读者更好地设计适用于特定DNA序列的引物。
引物的基本要求PCR引物应满足一定的要求才能确保PCR反应的成功。
以下是PCR引物的基本要求:1.引物长度通常为18-25个碱基对,过长或过短的引物均会影响PCR效率和特异性。
2.引物的GC含量通常在40%-60%之间,过高或过低的GC含量会影响引物的稳定性。
3.引物的3’末端应尽量避免G或C碱基,以减少引物之间的互补性。
4.引物之间的Tm(退半温度)差异最好在2℃以内,以确保引物能够同时结合在目标DNA序列上。
5.引物之间不应有明显的互补性,以免引起非特异扩增。
6.引物与非目标DNA序列的互补性应尽量避免,以确保特异性扩增。
PCR引物设计方法以下是一种常用的PCR引物设计方法:1.根据目标DNA序列,选择PCR引物的起始位置。
一般选择在目标序列两侧约200-300个碱基对的位置。
确保引物区域内无重复序列,避免非特异扩增。
例:目标DNA序列为ACGTAGCTAGCTAGC欲设计的引物起始位置为第5个碱基对,选取引物区域为AGCTAGCTAGC2.对引物区域进行BLAST查询,确保引物区域在目标DNA序列中没有互补序列。
例:通过BLAST查询,确认引物区域无互补序列3.根据引物区域设计引物的前向引物和反向引物。
引物长度通常为18-25个碱基对,GC含量应在40%-60%之间,3’末端避免使用G或C碱基。
例:前向引物为AGCTAGCTAGCGACGCGC反向引物为GCGCGTCGCTAGCTAGCT4.使用引物设计软件或在线工具(如NCBI Primer-BLAST)进行引物性能评估。
输入引物序列,并检查引物间的Tm差异是否在2℃以内,检查引物之间以及与非目标序列的互补性。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
P C R引物流程设计详
解
PCR引物设计流程详解
本文目的:复制出IL-4基因片段
一、查找基因序列
1、进入NCBI主页,下拉选框选择Nucleotide,在搜索栏输入要查找的目
的基因,即IL-4,点击搜索
2 、在搜索结果选择灵长类(Homo sapiens)
2、在灵长类IL-4基因中选择需要的mRNA序列
3、查看基因的相关信息
外显子区域
CDs区域
4、点击FASTA格式,并将序列保存到文档
二、使用primer premier 5.0设计引物
1、建立新文件,将所得的序列复制进输入框内
2、点击搜索按钮,搜索引物
3、设置引物设计参数(因为在之前查找基因序列的时候获知,外显子区域
分别为:1-200、201-248、249-425、426-618,又知在引物设计时引物位置最好跨越一个内含子,PCR产物长度通常为100-150bp,故设定上游引物位置为201-248,下游引物位置为249-425,产物长度为100-150bp)
4、确认条件后,显示搜索结果
4、双击选中得分最高的引物查看引物情况
(上图为上游引物情况,下图为下游引物情况)
5、将设计的上下游引物复制出来,保存到文档中
三、使用oligo 6.0对设计的引物进行评价
1、建立新文件,将从cnki上获得的cDNA复制进输入框,并点击accept接
收
2、接收后显示出该序列的相关信息
3、点击edit按钮录入用primer设计的上游引物,每一次输入新数据后都
需要点击accept按钮接收
4、同理,录入下游引物
5、分析上下游引物二聚体形成情况
6、分析上下游引物发卡形成情况
7、分析上下游引物GC%
8、检测上下游引物与PCR模板其它位置错配情况
9、分析PCR整体情况
四、引物特异性检验(primer blast)
1、进入NCBI主页,并选择blast
2、选择primer blast
3、在输入框内输入模板序列和上下游引物,并设定对比数据库,点击get
primer进行对比
4、查看blast结果
Blast 结果显示,尽管IL-4与其它基因有相似区,但是引物的3’端没有完全互补。
故设计的引物能特异性的扩增出目的基因。