完全型雄激素不敏感综合征20例临床分析_董晓超
完全性雄激素不敏感综合征遗传病学探究

磺士避文结果一、临床资料2名患者具有正常女性外观的外生殖器,有睾丸(隐睾,位于腹股沟和腹腔),无月经,核型为46,xY,临床诊断为完全性雄激素不敏感综合征。
4名家族成员经过体格检查,辅助检查和实验室检查初步认定为正常女性。
二、PER及基因测序结果(1)2例CAIS患者的雄激素受体(AR)基因2号外显子缺失(见图2-1)。
其余外显子电泳条带存在(见图2—2),经过基因测序与正常AR基因相符。
123456(图2一lARExon2PcR产物凝胶电泳成像)注:编号1、2为2名患者,3为患者母亲,4-6为家族成员E2E3E4E5E6E7(图2-2患者AR基因外显子检测结果)磺士避童(Exon7测序结果)(2)其中一位患者母亲的雄激素受体基因2号外显子PCR产物凝胶电泳成像后条带暗淡,提示其为该致病基因的携带者(见图2-1箭头所指)。
18硬士论文(3)AIR基因的Exon2及相邻内含子区域碱基序列分析:2例CAIS患者2号外显子两侧的内含予有缺失,Exon2上游2250bp至下游3785bp内含子缺失。
根据正常AR基因设计引物PCR扩增Exon2上下游1400bp及4000bp长度的片段在凝胶电泳中均未成像(见图2.3)。
在Exon2的5’端距Exon21700bp、2000bp、2250bp和2600bp处分别设计引物,将PCR扩增所得产物凝胶电泳,并与正常AR基因比较,发现1700bp、2000bp和2250bp处未成像,而2600bp处电泳条带与『F常AR基因相符(见图2—4)。
在Exon2的3’端距Exon23569bp、3785bp、4165bp、5230bp、6103bp和7202bp分别处设计引物对,将PCR扩增所得产物凝胶电泳,并与正常AR基因比较,发现3569bp、3785bp处未成像,而4165bp、5230bp、6103bp和7202bp处的电泳条带与正常AR基因相符(见图2.5、2-6)。
雄激素不敏感综合征的诊断和鉴别诊断

诊断-病史
– 按女性生活的患者有原发闭经,可有腹 股沟或阴唇内包块,部分患者出生后有 外生殖器性别模糊,到达青春期后有阴 蒂增大或男性化表现
AR基因突变
• 缺失型:仅占5~10%,包括整个基因或部分基因 的丢失或3的非整倍数碱基的缺失
• 点突变型:最为常见,可导致:
–形成提前终止密码,导致受体合成不完整,截短的受体 在与激素的结合和基因转录的激活方面是没有活性的
–剪切位点发生突变,引起mRNA的拼接异常 –点突变引起氨基酸替换,最为常见,约85%发生在甾体
完全型雄激素 不敏感综合征
+ + - 女性 盲端 无 无 无 睾丸 46,XY 正常或升高 正常或升高 无 无
46,XY 单纯性腺 发育不全
+ - - 女性 有 有 有 有 条索状 46,XY 低下 低下 无 无
46,XY 17α 羟化酶缺乏
+ - - 女性 盲端 无 无 无 睾丸 46,XY 低下 低下 有 有
CAIS T 升高 4 正常 13 低下 6
E2 LH FSH PRL 4 22 16 - 13 1 6 12 4 -- 1
田秦杰,刘慧,郎景和。中国实用妇科与产科杂志。2004,20(12):723-725
AIS睾酮水平低下的可能原因
• 长期升高的LH可能导致睾丸间质细胞的 LH受体下降,即间质细胞LH受体发生下 调节,引起睾酮继发性合成障碍?
• 对那些AR结合质量异常和对人工合成的雄 激素类药物有反应的,在超生理剂量或改 变雄激素类型后,雄激素效应将可达到正 常男性水平,这类患者在新生儿和青春期 给予治疗仍可按男性生活
雄激素不敏感综合征

雄激素不敏感综合征定义:由于雄激素靶器官上的雄激素受体(Androgen Receptor,AR)出现障碍而导致对雄激素不反应或反应不足而导致多种临床表现,称为雄激素不敏感综合征(Androgen Insensitivity Syndrome,雄激素不敏感综合征),以往曾被称为“睾丸女性化(Testicular feminization)”,现已不用此名称。
分类:以往根据临床表现,将雄激素不敏感综合征详细地分为五类,即:1. 完全型睾丸女性化2. 不完全型睾丸女性化3. Reifenstein综合征(会阴阴囊型尿道下裂)4. 男性不育综合征5. 男性化不足综合征但仅前三类有明显的性发育异常,故目前临床上根据患者有无男性化表现,将雄激素不敏感综合征患者分为两大类,即:1. 无男性化表现的完全型雄激素不敏感综合征(Complete AIS,CAIS):即完全型睾丸女性化。
2. 有男性化表现的不完全型或部分型雄激素不敏感综合征(Incomplete或Partial AIS,IAIS或P AIS):即不完全型睾丸女性化和Reifenstein综合征。
发生率:雄激素不敏感综合征占原发闭经的6-10%,发病率为出生男孩的1/20,000-64,000。
在儿科有腹股沟疝而手术的“女孩”中,雄激素不敏感综合征的发生率为1.2%。
病因:受累的雄激素不敏感综合征个体遗传上为男性,因为其核型为46,XY,有睾丸。
患者的血浆睾酮、双氢睾酮与雌激素水平均在男性正常范围。
用hCG刺激后,雄激素与雌激素水平上升,说明性激素均来自睾丸且反应正常。
其病因是雄激素靶器官上的雄激素受体出现障碍而导致雄激素的正常效应全部或部分丧失。
雄激素必须通过雄激素受体才能起作用。
雄激素受体是一种配基(雄激素)依赖性转录因子,与糖皮质激素、盐皮质激素、孕激素、雌激素、维生素D3和甲状腺素等受体同属一类,有类似的结构。
雄激素受体是一种对雄激素有高亲和力的结合蛋白,通过诱导靶基因的转录,而介导睾酮和双氢睾酮的生理效应。
雄激素不敏感综合征临床诊治分析(附6例报告)

( De p a r t me n t o f Cl i n i c a l Me d i c i n e ,Me d i c a l S c h o o 1 ,S o o c h o w Un i v e r s i t y, S u z h o u ,J i a n g s u, 2 1 5 0 0 6,Ch i n a ; 。 De p a r t me n t o f Ur o l o g y ,S i x t h P e o p l e Ho s p i t a l , S h a n g h a i J i a o t o n g Un i v e r s i t y )
Co r r e s p o nd i ng a u t ho r :SA Yi ng l o ng,E— ma i l :s a y i ng l o ng 33 1( s i na .e o m Ab s t r a c t Ob j e c t i v e : Ob j e c t i v e To i n v e s t i g a t e t h e d i a g n o s i s a n d t h e t r e a t me n t o f a n d r o g e n i n s e n s i t i v i t y s y n —
d r o me pa t i e nt s t O i mp r ov e t he e f f i c i e n c y. Me t ho d: A r e t r o s p e c t i ve a na l y s i s we r e pe r f o r me d wi t h t ot a l of s i x c a s e s of
完全型雄激素不敏感综合征1例并文献复习
完全型雄激素不敏感综合征1例并文献复习发布时间:2021-05-14T10:19:45.803Z 来源:《中国医学人文》2021年9期作者:熊蕊,耿伟,王斐,王爱敏,通讯作者:王烈宏[导读] 雄激素不敏感综合征(AIS)是46,XY性发育障碍(DSD)中最常见的类型熊蕊,耿伟,王斐,王爱敏,通讯作者:王烈宏青海红十字医院妇产科,青海西宁810000摘要:雄激素不敏感综合征(AIS)是46,XY性发育障碍(DSD)中最常见的类型,发病率为1/10万~ 1/9万。
根据雄激素受体(AR)的量,AIS主要分为三类:完全型、部分型和轻度型。
完全型雄激素不敏感综合征是(CAIS)最严重的一种类型,核型为46,XY,却表现出完全的女性外生殖器表型及女性第二性征。
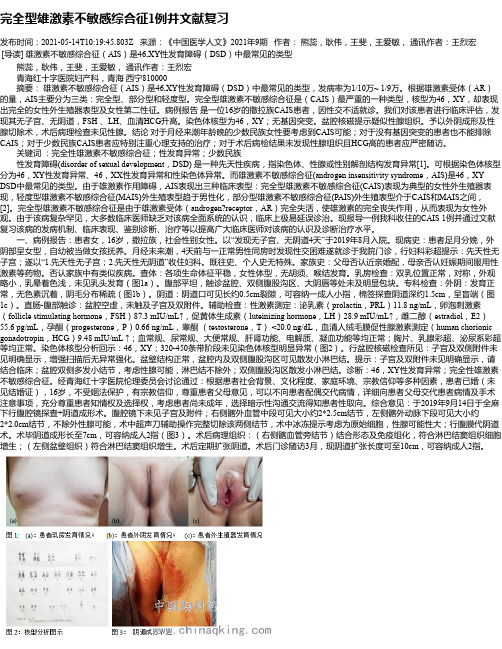
病例报告是一位16岁的撒拉族CAIS患者,因性交不适就诊。
我们对该患者进行临床评估,发现其无子宫、无阴道,FSH 、LH、血清HCG升高。
染色体核型为46,XY;无基因突变。
盆腔核磁提示疑似性腺组织。
予以外阴成形及性腺切除术,术后病理检查未见性腺。
结论对于月经来潮年龄晚的少数民族女性要考虑到CAIS可能;对于没有基因突变的患者也不能排除CAIS;对于少数民族CAIS患者应特别注重心理支持的治疗;对于术后病检结果未发现性腺组织且HCG高的患者应严密随访。
关键词:完全性雄激素不敏感综合征;性发育异常;少数民族性发育障碍(disorder of sexual development,DSD) 是一种先天性疾病,指染色体、性腺或性别解剖结构发育异常[1]。
可根据染色体核型分为46,XY性发育异常、46,XX性发育异常和性染色体异常。
而雄激素不敏感综合征(androgen insensitivity syndrome,AIS)是46,XY DSD中最常见的类型。
由于雄激素作用障碍,AIS表现出三种临床表型:完全型雄激素不敏感综合征(CAIS)表现为典型的女性外生殖器表现,轻度型雄激素不敏感综合征(MAIS)外生殖表型趋于男性化,部分型雄激素不敏感综合征(PAIS)外生殖表型介于CAIS和MAIS之间,[2]。
2024儿童雄激素不敏感综合征诊断和治疗专家共识(全文)

2024儿童雄激素不敏感综合征诊断和治疗专家共识(全文)摘要雄激素不敏感综合征(AIS)是46,XY性发育异常疾病常见病因之一,根据雄激素受体功能的缺陷程度可分为完全型、部分型及轻微型。
儿童AIS 的内分泌学特征为持续性雄激素抵抗,其临床管理的核心在于性别认同,改善青春期性发育特征。
现以国内外研究为基础,结合国内专家的临床诊疗经验,在AIS的遗传学分子机制、临床表现、诊断及鉴别诊断、抚养性别评估、肿瘤风险、内分泌激素及外科手术治疗时机、长期预后等方面形成系统、全面的诊断和治疗专家共识,为儿科医师提供规范的临床诊治指导。
雄激素不敏感综合征(androgen insensitivity syndrome,AIS)是由于雄激素受体(androgen receptor,AR)基因变异导致雄激素受体功能缺陷,虽然雄激素合成正常,但靶器官对雄激素作用无应答(抵抗)或应答不足,从而出现不同程度男性化不全。
该病为一种X连锁隐性遗传病,属于46,XY 性腺发育异常疾病(disorders of sex development,DSD)常见的类型之一[1]。
AIS临床表型依赖于雄激素受体功能缺陷程度,分为3种类型:完全型AIS(complete AIS,CAIS)、部分型AIS(partial AIS,PAIS)和轻微型AIS(mild AIS,MAIS)。
国外报道CAIS发病率为1/99 000~1/20 400[2],PAIS发病率为1/130 000[3, 4]。
国内报道AIS发病率仅次于5α还原酶缺乏症[5]。
AIS在抚养性别的共同决策、性腺处理及切除时机的选择、肿瘤风险的监测以及性激素的合理替代等方面尚缺乏共识,需要发挥多学科合作,是儿童内分泌科医生面临的重大挑战。
现为提高AIS诊疗水平,中华医学会儿科学分会内分泌遗传代谢学组、中华儿科杂志编辑委员会、国家儿童健康与疾病临床医学研究中心组织相关专家,经过多次研讨会,历时1年余,制订儿童AIS诊断和治疗专家共识(以下简称本共识)。
雄激素不敏感综合征临床诊断及治疗:附七例病例分析

京妇产 医院 1 9 9 7 -2 0 1 2年收治的 7例 A I S患者的临床 资料 ,对疾病的诊 断及 治疗进行总结。结果 瘤 ,术后均 口服 方 式 可 以提 高 患 者 生 活质 量 、 减 少肿 瘤发 生 。
不 同程度女性 型,染色体核型为 4 6 ,X Y,5例患者睾酮水平 高于正常 男性 。7例 均行 手术切 除睾丸 ,5例合并睾 丸肿
B e i j i n g 1 0 0 0 2 6,C h i n a
【 A b s t r a c t 】
0b j e c t i v e T o a n a l y z e t h e c h a r a c t e i r s t i c s o f t h e d i a g n o s i s a n d t r e a t me n t o f a n d r o g e n i n s e n s i t i v i t y s y n d r o m e
原发 闭经患者睾 酮为男性水平 、染 色体核型 为 4 6 ,X Y时
应考虑为 A I S ,青春期前的患者应行人绒毛膜促 性腺 激素 ( H C G) 试验 了解性腺 功能。根据 A I S分类选择手 术时机 及 【 关键 词】 雄激素迟钝 综合征 ;诊断 ;治疗
【 中图分类号 】R 5 8 8 . 1 【 文献标识码 】B d o i :1 0 . 3 9 6 9 / j . i s s n . 1 0 0 7 — 9 5 7 2 . 2 0 1 5 . 3 5 . 0 2 6
・
437 8・
・
临床 诊 疗 提 示 ・
雄 激 素 不 敏 感 综 合 征 临 床 诊 断 及 治 疗 : 附 七 例 病 例 分 析
完全型雄激素不敏感综合征1例

完全型雄激素不敏感综合征1例作者:黄晓娟何菊仙蔡华来源:《医学信息》2020年第16期关键词:雄激素不敏感综合征;遗传病;假两性畸形雄激素不敏感综合征(AIS)是雄激素受体基因失活或突变引起的罕见的X-连锁隐性遗传病,染色体核型为46XY,属于男性假两性畸形,发病率为出生男孩的(1.6~5.0)/10万[1]。
患者临床表现为一系列雄激素抵抗综合征,根据机体对雄激素抵抗严重程度分为完全型雄激素不敏感综合征(CAIS),部分型雄激素不敏感综合征(PAIS),轻微型雄激素不敏感综合(MAIS),其表型从类似于女性外生殖器到正常男性表型仅伴有不育或乳房发育等。
我院2019年收治了1例CAIS患者,现将诊治经过报道如下,以期为該疾病的诊治提供参考。
1临床资料患者,女,22岁,未婚,因“发现生殖器发育异常3年余”于2019年6月12日就诊于我院。
患者青春期后月经未来潮,于19岁就诊于当地医院查B超提示:先天性无子宫,右侧阴唇皮下及左侧腹股沟区可见睾丸回声。
之后未于进一步诊治。
现就诊于我院行超声检查示:先天性无子宫,右侧阴唇皮下及左侧腹股沟区可见睾丸回声。
染色体检查示:常染色体44,性染色体2,核型XY。
无腹胀、腹痛及特殊不适。
患者系父母于17 d大小时抱养,原生家庭情况不详。
既往体健,否认高血压、低血钾等特殊病史。
查体:生命体征平稳,女性体态,身高:167 cm,体重:49 kg。
无喉结,乳房发育饱满,乳头偏小,腋毛稀疏;外生殖器检查:外阴,阴毛少,呈女性分布,阴唇发育可,舟状窝下缘可探及一约2 cm盲段,宽约1 cm,尿道开口位于其中,右侧大阴唇皮下可扪及一约4 cm×3 cm×;;; 3 cm椭圆形包块,活动好,无压痛。
左侧腹股沟区可扪及一约4 cm×3 cm×3 cm椭圆形包块,活动好,无压痛。
2019年6月14日磁共振(盆腔平扫):膀胱直肠间未见子宫及阴道信号,盆腔未见确切卵巢信号。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
文章编号:1003-6946(2015)01-070-03完全型雄激素不敏感综合征20例临床分析董晓超1,金杭美2(1.浙江省杭州市第一人民医院,浙江杭州310006;2.浙江大学医学院附属妇产科医院,浙江杭州310006)【摘要】目的:探讨完全型雄激素不敏感综合征的临床特征与诊治策略。
方法:回顾性分析20例完全型雄激素不敏感综合征患者的临床资料。
结果:20例患者均为女性表型但染色体核型为46,XY,乳房发育正常却无月经来潮,腋毛和阴毛稀少,阴道呈短浅盲端,子宫缺如,性腺位于盆腔、腹股沟管或大阴唇内。
所有患者均行预防性性腺切除术,其中5例同时行腹股沟疝修补术,3例同时行大阴唇皮瓣移植阴道成形术。
术后性腺病理诊断均为睾丸组织,其中2例局部呈错构瘤样增生(Pick腺瘤),2例伴支持细胞腺瘤样结节形成,4例尚有发育不良的输卵管组织。
结论:完全型雄激素不敏感综合征的临床诊断需结合症状体征、性激素水平、影像学资料及染色体核型等方面综合考虑。
对于女性社会性别的患者,治疗的关键在于适时的睾丸切除和必要的阴道重建。
【关键词】完全型雄激素不敏感综合征;睾丸切除;阴道重建中图分类号:R588文献标志码:B雄激素不敏感综合征(androgen insensitivity syn-drome,AIS)又称睾丸女性化综合征(testicular femini-zation syndrome,TFS),是男性假两性畸形中的常见类型,首先由John Morris于1953年报道并命名,而后根据雄激素不敏感程度又分为完全型(complete andro-gen insensitivity syndrome,CAIS)、部分型(partial an-drogen insensitivity syndrome,PAIS)及轻微型(mild an-drogen insensitivity syndrome,MAIS)3类,其中以CAIS 较多见,发病率占出生男婴的1/64000 1/20000[1]。
现总结20例CAIS患者的临床资料,对CAIS的临床特征及诊治策略进行分析探讨。
1临床资料浙江大学医学院附属妇产科医院2008年7月至2013年6月收治20例CAIS患者,年龄16 34岁,平均21.6ʃ5.6岁;身高158 178cm,平均164.3ʃ6.3cm;体重42 75kg,平均56.2ʃ9.2kg。
4例已婚,16例未婚(其中4例有性生活)。
12例因原发性闭经首诊,8例曾因原发性闭经就诊发现生殖道畸形但未进一步诊治,其中4例因腹股沟包块复诊,3例因性生活障碍复诊,1例因大阴唇包块复诊。
家族史均无异常。
2结果2.1临床表现20例患者均为女性表型,无胡须与喉结,腋毛稀少,乳房发育良好,乳晕色浅,乳头偏小。
女性外阴,阴毛少或无,阴蒂与阴唇发育正常或偏小,阴道呈盲端,宽可容1 2指,长1 9cm,平均5.2ʃ2.1cm,盆腔空虚,未触及子宫。
其中4例于双侧腹股沟区、1例于双侧大阴唇内可触及实性肿物,直径2 3cm,质偏软,边界清,活动可,无压痛。
2.2辅助检查20例患者超声检查提示盆腔内未见子宫及卵巢样回声,在盆腔腹股沟管上方(10例)、盆腔髂血管旁(5例)、腹股沟区(4例)或大阴唇内(1例)可见中等或偏低回声包块,边界清,内有少量血流信号。
血清性激素水平测定结果见表1。
外周血染色体核型为46,XY。
2.3诊断根据临床表现特征与辅助检查结果,20例患者均诊断为CAIS。
性腺病理诊断均为睾丸组织,未见生精细胞,其中2例局部呈错构瘤样增生(Pick 腺瘤),2例伴支持细胞腺瘤样结节形成,4例尚有发育不良的输卵管组织。
[22]刘小艳,常青.早孕期终止瘢痕子宫患者意外妊娠的临床分析[J].重庆医学,2012,41(8):759-761.[23]Shao MJ,Hu MX,Xu XJ,et al.Management of caesarean scar preg-nancies using an intrauterine or abdominal approach based on the myometrial thickness between the gestational mass and the bladder wall[J].Gynecol Obstet Invest,2013,76(3):151-157.[24]Ficicioglu Cem,AttarR,Yildirim Gazi.Fertility preserving surgicalmanagement of methotrexate-resistant cesarean scar pregnancy[J].Taiwan J Obstet Gynecol,2010,49(2):211-213.[25]Zhang Y,Duan H,Cheng JM,et al.Treatment options to terminate,persistent cesarean scar pregnancy[J].Gynecol Obstet Invest,2013,75(2):115-119.(收稿日期:2014-08-25;修回日期:2014-11-01)表120例CAIS患者血清性激素水平测定结果测定值参考值(本院正常女性)参考值(本院正常男性)LH(U/L)26.93ʃ16.01(4.12 69.72) 3.8 20.0 2.2 8.4FSH(U/L)20.81ʃ19.31(2.31 63.77) 3.8 17.2 2.3 9.5T(nmol/L)21.96ʃ10.47(10.10 43.80)0.3 3.0 6.6 35.0E2(pmol/L)140.96ʃ60.12(55.75 300.30)92.0 275.0①;367.0 1100.0②28.0 156.0P(nmol/L) 2.09ʃ0.78(1.17 4.09)0.3 4.8①;8.0 89.0②—①卵泡期;②黄体期2.4治疗所有患者均行预防性双侧性腺切除术,术后每日口服戊酸雌二醇片(补佳乐)1mg或结合雌激素片(倍美力)0.625mg长期替代治疗。
5例存在腹股沟疝者同时行疝修补术。
8例有性生活者,3例阴道仅为深1 2cm的浅凹,因性生活障碍同时行大阴唇皮瓣移植阴道成形术;其余5例阴道长6 9cm,予保守观察。
12例未婚者阴道长3 7cm,因无性生活需求未作特殊处理,建议婚前评估,必要时再行阴道重建。
2.5随访20例患者术后随访3 12个月,女性第二性征及心理状态均保持良好,其中5例腹股沟疝修补术后患者未复发,3例阴道成形术后患者坚持佩戴阴道模具,阴道无挛缩狭窄、瘢痕粘连,性生活质量明显改善。
3讨论3.1发病机制AIS是一种与雄激素受体编码基因突变密切相关的疾病,目前已知的突变类型超过800种[2]。
患者染色体核型为46,XY,使胚胎性腺发育成睾丸并分泌雄激素,但雄激素受体却因基因突变而缺失或减少,导致靶器官对雄激素不敏感(雄激素抵抗),雄激素的生物学效应完全或部分丧失,睾丸下降受限成为隐睾,中肾管发育障碍难以形成附睾、精囊及输精管;而睾丸分泌的副中肾管抑制因子(MIF)又能阻碍子宫、输卵管及阴道上部的分化,从而在睾丸分泌的雌激素与雄激素外周转化的雌激素共同作用下最终形成不同程度的女性化外观与外生殖器表型。
研究还发现,CAIS中70%是X连锁隐性遗传病,通过女性携带者遗传给后代,后代中女性50%为携带者,男性50%患病,另有30%为基因新生突变所致[3]。
3.2诊断及临床表现目前对AIS的研究已深入到分子水平,但其临床诊断尚需结合症状体征、性激素水平、影像学资料及染色体核型等方面综合考虑,并排除先天性无阴道综合征与单纯性腺发育不全综合征引起的原发性闭经、先天性肾上腺皮质增生导致的男性化现象以及其他原因造成的男性假两性畸形,有条件者还可取外生殖器皮肤成纤维细胞培养测定雄激素结合能力,并应用分子生物学技术鉴定基因突变明确病因学诊断[4]。
AIS的临床表现变异性较大,患者常因原发性闭经、生殖器畸形、腹股沟或大阴唇肿物就诊。
其体征的多样性取决于雄激素不敏感的程度,临床上据此将AIS分为CAIS、PAIS及MAIS3类[5,6]:CAIS患者的女性化程度高,平均身高高于正常女性但低于正常男性水平;无胡须与喉结,乳房发育较好,但通常乳头偏小、乳晕色浅,腋毛和阴毛稀疏或缺如;外阴为女性型,阴道呈窄盲端,短者仅表现为前庭浅凹,长者可接近正常阴道长度,一般无子宫与输卵管;睾丸可位于腹盆腔、腹股沟管或大阴唇等睾丸下降途径中的任何部位。
血清性激素水平测定显示睾酮(T)与卵泡刺激素(FSH)多在正常或高于正常男性水平,雌二醇(E2)高于正常男性但相当于女性卵泡期水平,黄体生成素(LH)水平因睾酮对下丘脑-垂体系统的负反馈异常而升高。
睾丸病理检查可见大量无生精功能的曲细精管,无附睾和输精管。
相比之下,PAIS患者因雄激素受体活性部分存留而具有不同程度的男性化表现,如有胡须与喉结、声音增粗、皮肤粗糙,外生殖器形成从类似正常女性到接近正常男性的广泛表型,如阴蒂肥大、阴唇融合、尿道下裂。
MAIS患者因仅有轻微的雄激素抵抗而呈男性表型,在青春期后出现男性化不足,如乳房发育、阴茎偏小、性毛稀疏、少精液症或男性不育。
本组患者根据其临床特征与手术结果均可确诊为CAIS。
值得注意的是,有4例患者的术后病理检查发现了理论上不应存在的输卵管组织。
以往也有学者发表过类似报道,认为CAIS患者可有发育不良的输卵管或子宫,且这种苗勒管残留现象或许比现有的报道更多,其原因可能与MIF分泌不足或功能缺陷、雌激素过多导致MIF作用减弱以及睾丸下降早期苗勒管结构不受MIF影响等有关[7]。
3.3治疗策略AIS患者出生后应根据外阴性别倾向与手术治疗预期选择合适的社会性别抚养,在生长发育期间给予必要的心理调节,并尽早重建外生殖器使之与抚养性别一致。
对于女性社会性别的CAIS患者,临床治疗的关键在于适时的睾丸切除和必要的阴道重建。
3.3.1睾丸切除CAIS患者的睾丸有发生肿瘤的风险,主要是睾丸发育异常导致原始生殖细胞成熟障碍,可在曲细精管内逐渐形成肿瘤前驱病变(小管内生殖细胞肿瘤未定类型/原位癌),继续发展可形成精原细胞瘤、性母细胞瘤等多种生殖细胞肿瘤;而持续高水平的黄体生成素还可使睾丸间质异常增生,形成支持细胞瘤、间质细胞瘤等非生殖细胞肿瘤[6,8]。
由于研究范围的限制与调查方法的差异,CAIS患者的睾丸肿瘤发病率尚无确切估计,以往报道在0.8% 22%,最近研究显示为14%[9],且在青春期前仅为0.8% 2%,至青春期后随年龄增长可高达30%以上[8],可见早期切除睾丸可预防肿瘤发生。