附双向电泳完整的操作步骤第一向等电聚焦1从冰箱中取-20
双向电泳详细操作过程
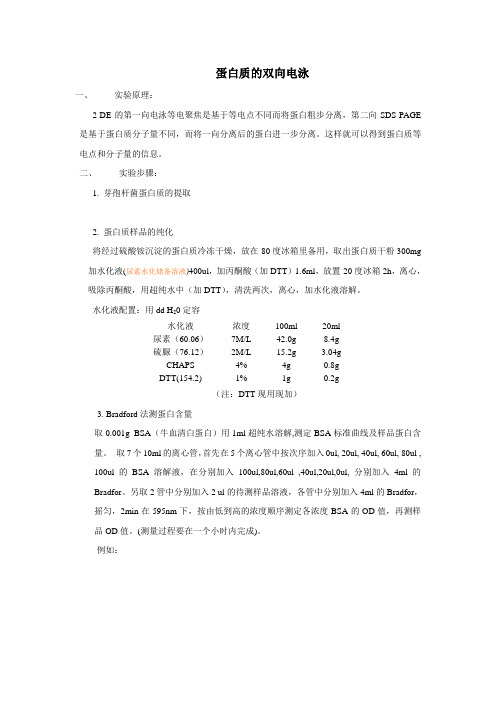
蛋白质的双向电泳一、实验原理:2-DE的第一向电泳等电聚焦是基于等电点不同而将蛋白粗步分离,第二向SDS-PAGE 是基于蛋白质分子量不同,而将一向分离后的蛋白进一步分离。
这样就可以得到蛋白质等电点和分子量的信息。
二、实验步骤:1. 芽孢杆菌蛋白质的提取2. 蛋白质样品的纯化将经过硫酸铵沉淀的蛋白质冷冻干燥,放在-80度冰箱里备用,取出蛋白质干粉300mg 加水化液(尿素水化储备溶液)400ul,加丙酮酸(加DTT)1.6ml,放置-20度冰箱2h,离心,吸除丙酮酸,用超纯水中(加DTT),清洗两次,离心,加水化液溶解。
水化液配置:用dd H20定容水化液浓度100ml 20ml尿素(60.06)7M/L 42.0g 8.4g硫脲(76.12)2M/L 15.2g 3.04gCHAPS 4% 4g 0.8gDTT(154.2) 1% 1g 0.2g(注:DTT现用现加)3. Bradford法测蛋白含量取0.001g BSA(牛血清白蛋白)用1ml超纯水溶解,测定BSA标准曲线及样品蛋白含量。
取7个10ml的离心管,首先在5个离心管中按次序加入0ul, 20ul, 40ul, 60ul, 80ul , 100ul的BSA溶解液,在分别加入100ul,80ul,60ul ,40ul,20ul,0ul, 分别加入4ml的Bradfor。
另取2管中分别加入2 ul的待测样品溶液,各管中分别加入4ml的Bradfor,摇匀,2min在595nm下,按由低到高的浓度顺序测定各浓度BSA的OD值,再测样品OD值。
(测量过程要在一个小时内完成)。
例如:标准曲线方程式:Y= aX+b.其中Y为OD值,X为蛋白含量。
a、b通过作图输入数据可知G250的配置:称取G250 固体0.1g加水定容至1L。
使用前滤纸过滤。
比色皿用70%的乙醇保存,待用时用双蒸水冲洗,再用无水乙醇冲洗,双蒸水冲洗,再加入待测样品溶液润洗,然后,加入样品,测定OD值。
简述双向电泳技术分离蛋白质的基本技术流程

简述双向电泳技术分离蛋白质的基本技术流程下载提示:该文档是本店铺精心编制而成的,希望大家下载后,能够帮助大家解决实际问题。
文档下载后可定制修改,请根据实际需要进行调整和使用,谢谢!本店铺为大家提供各种类型的实用资料,如教育随笔、日记赏析、句子摘抄、古诗大全、经典美文、话题作文、工作总结、词语解析、文案摘录、其他资料等等,想了解不同资料格式和写法,敬请关注!Download tips: This document is carefully compiled by this editor. I hope that after you download it, it can help you solve practical problems. The document can be customized and modified after downloading, please adjust and use it according to actual needs, thank you! In addition, this shop provides you with various types of practical materials, such as educational essays, diary appreciation, sentence excerpts, ancient poems, classic articles, topic composition, work summary, word parsing, copy excerpts, other materials and so on, want to know different data formats and writing methods, please pay attention!双向电泳技术分离蛋白质双向电泳技术是一种常用于分离复杂蛋白质混合物的方法。
双向电泳操作步骤

双向电泳操作步骤双向电泳操作步骤及相关溶液配置A(实验过程一实验原理:2-DE的第一向电泳等电聚焦是基于等电点不同而将蛋白粗步分离,第二向SDS-PAGE是基于蛋白质分子量不同,而将一向分离后的蛋白进一步分离。
这样就可以得到蛋白质等电点和分子量的信息。
二实验步骤:1. 样品的溶解取纯化后的晶体蛋白3.0mg,加入300ul裂解液(1mg蛋白:100ul裂解液)振荡器上振荡10min左右,共处理一个小时。
其中每隔10,15分钟振荡一次,然后13200rpm离心15min除杂质,取上清分装,每管70ul,—80oC保存。
2. Bradford法测蛋白含量取0.001g BSA(牛血清白蛋白)用1ml超纯水溶解,测定BSA标准曲线及样品蛋白含量。
取7个10ml的离心管,首先在5个离心管中按次序加入0ul, 5ul,10ul, 15ul, 20ul 的BSA溶解液,另2管中分别加入2 ul的待测样品溶液,再在每管中加入相应体积的双蒸水(总体积为80ul),然后,各管中分别加入4ml的Bradford液(原来配好的Bradford液使用前需再取需要的剂量过滤一遍方能使用),摇匀,2min在595nm下,按由低到高的浓度顺序测定各浓度BSA的OD值,再测样品OD值。
(测量过程要在一个小时内完成)。
3. 双向电泳第一向---IEF(双向电泳中一律使用超纯水)3.1 水化液的制备称取2.0mg 的DTT,用700ul水化液储液溶解后,加入8ul 0.05, 的溴酚兰,3.5ul(0.5,v/v)IPG buffer (pH 3-10)振荡混匀,13200rpm离心15min 除杂质,取上清。
在含300ug 蛋白(经验值)的样品溶解液中加入水化液,至终体积为340ul,振荡器上振荡混合,13200rpm离心15min除杂质,取上清。
3.2 点样,上胶分两次吸取样品,每次170ul, 按从正极到负极的顺序加入点样槽两侧,再用镊子拨开 Immobiline DryStrip gels (18cm,pH 3—10)胶条,从正极到负极将胶条压入槽中,胶面接触加入的样品。
双向电泳实验操作流程

双向电泳实验操作流程机器型号:GE第一向:等电聚焦(IEF)1.对样品的要求:IEF能否成功主要取决于样品状态和离子强度,一般蛋白制备采用Trin-HCl 为缓冲液,加等渗蔗糖。
根据染色方法和胶条的长度决定上样量,一般是20μg/mL -1mg/mL。
考马斯亮蓝染色较银染需要较大的上样量。
2.IEF准备注意事项:DTT和IPG现用现配,由于DTT是还原剂,长时间放置易氧化,所以可以选择干物质使用。
DTT的作用是和尿素配合打散蛋白质结构,CHAPS为碱性去垢剂,和SDS作用相似,溶解尿素时温度不可超过37℃,因为超过37℃蛋白质会发生氨甲酰化,尽量在生产日期一年内使用。
清洗IEF胶条槽时用专用清洗剂,原液清洗或2%浓度浸泡清洗。
3.上样:胶条使用前20min从冰箱(4℃)中取出。
上样可以采用水化上样或者使用上样杯(先水化,后上样),上样杯上样适用于极性等电点蛋白质,水化12h后,在电泳槽上样。
以水化上样为例:先将250μL样品和水化液(需要当天配制)混合物加到胶条槽里,然后从尖端(阳性端)撕掉保护膜(带IO号),将胶条支持膜向上,胶面向下,用配用镊子夹住平端放入胶条槽中,注意不要产生气泡,用覆盖油覆盖,盖上盖子。
说明:放入胶条槽中的胶条上的字应该顺向能够读出,说明胶条放的对,如果读不出来,说明放反了。
在电脑软件中设置操作参数:水化上样分为被动上样(就是这种浸泡状态维持约16h)和主动上样(加电压30V,大约需要10h)。
上样后即可进行IEF电泳。
注意每种胶条最大的电流不能超过50μA。
一般是30V电压水化12h,然后200V、500V、1000V各1h,最后用8000V电压电泳至结束。
4.胶条的平衡:平衡液制备时先将SDS在加热的条件下溶解于水中,SDS溶解后冷却至20℃左右,在加入尿素充分溶解,然后加甘油混匀成水溶液。
将该水溶液分成两份,每份15mL,分装到两个平衡管中,其中一个管中加DTT,另一个管中加IAA,DTT可以打开蛋白的二硫键,防止在聚焦时形成的氧化导致在第二向拖尾,尿素、甘油可以降低电离效应,使胶条可以很好的转入第二向,IAA可以将去除DTT。
双向电泳操作流程

双向电泳完整操作流程仪器:Eppendorf 冷冻离心机 (Eppendorf)、Beckman Coulter 高速冷冻离心机(Beckman Coulter)、IPGhor 等电聚焦仪(GE Healthcare)、DALT-SIX SDS-PAGE电泳仪(GE Healthcare)、ImageScanner扫描仪(GE Healthcare)、ImageMaster 2D Platinum 7.0分析软件(GE Healthcare)、电子天平、分光光度计、旋涡混和器、PH计、真空冷冻干燥机、液氮、离心管、研钵主要溶液配置:三氯乙酸-丙酮沉淀液:10%三氯乙酸、0.07%巯基乙醇溶于100%丙酮丙酮洗涤液:0.07%巯基乙醇溶于丙酮样品裂解液:9mol/L尿素、4%CHAPS、1%IPG buffer(GE Healthcare)、1%DTT样品水化液:9mol/L尿素、4%CHAPS、1%IPG buffer(GE Healthcare)、1%DTT、少量溴芬兰定量染色液:0.01%(w/v)G250,8.5%磷酸和4.75%乙醇标准蛋白溶液:1mg/ml牛血清蛋白平衡缓冲液1:6mol/L尿素、50mmol/L Tris-HCL(pH=8.8)、30%甘油、2%SDS,1%DTT,痕量溴酚兰平衡缓冲液2:6mol/L尿素、50mmol/L Tris-HCL(pH=8.8)、30%甘油、2%SDS,4%碘乙酰胺,痕量溴酚兰12%SDS-PAGE凝胶溶液:12%丙烯酰胺、0.32%双丙烯酰胺、0.375mol/L Tris-HCL(pH=8.8)、0.1%SDS、0.05%过硫酸铵、0.05%TEMED 电泳缓冲液:25mmol/L Tris、192mmol/L甘氨酸、0.1%SDS封胶液:25mmol/L Tris、192mmol/L甘氨酸、0.1%SDS、0.5%琼脂糖考染固定液:12%(W/V)三氯醋酸考染染色液:0.12%G-250、10%(NH4)2SO4、10% H3PO4、20%甲醇。
双向电泳使用说明

第一部分介绍1.0手册的介绍这本手册分五大部分。
第一部分对手册进行了介绍。
第二部分介绍了样品预处理的方法。
第三部分细叙了进行双向电泳第一向电泳的过程。
第四部分介绍了利用IPG胶条进行第二向电泳的一般方法。
第五部分讨论了双向电泳的显影和结果分析。
在这里,使用Amersham pharmacia Bioteeh公司的产品进行双向电泳介绍,设备的选择我们在1、2节中介绍。
1.1 双向电泳的介绍双向电泳是分析从细胞、组织或其它生物样品中提取出来的蛋白混合物最有力和广泛运用的方法。
这项技术利用蛋白质在两次独立的分离步骤中的特性将蛋白质分开:第一向步骤——等电聚焦(IEF)根据蛋白质的等电点(PI)将蛋白质分离。
在第二向步骤中SDS—聚丙烯酰胺凝胶电泳(SDS—PAGE)利用蛋白质的分子量(MW)大小将它们分离。
二向电泳所得结果的斑点序列都对应着样品中的单一蛋白。
因此,上千种蛋白质均能被分离开来,并且各种蛋白质的等电点,分子量和含量的信息都能得到。
双向电泳在1975年由P.H.OFarrel[1]和J.Klose[2]提出,在早先的技术中,第一向分离在含有载体两性电解质的聚丙烯酰胺凝胶载体中进行,这种胶在狭窄的试管中灌注而成。
样品放入管胶的某一端,并且在很高的电压下将它们分开。
在完成等电聚焦(IEF)以后,凝胶棒从管子中取出来,在SDS样品缓冲液中平衡。
随后,放在垂直SDS—聚丙烯酰胺凝胶上,进行第二向分离。
自从双向电泳被被提出以来,双向电泳作为生化分离技术的重要性事实上早已被承认。
但是,它是在最近几年被广泛应用的,由于在各方面的改进。
■2—D技术被改进而产生了2—D的图象,这在分辨率和重复性上有很大提高。
这项新技术是由A.Gorg和他的同事发明的。
在2—D技术中,使第一向电泳得到了改进,利用固定的pH梯度代替了载体两性电解质产生的pH梯度,并且用塑料胶片支撑的凝胶条代替了柱状凝胶。
在章节3.1“Background to IEF”中阐述了这种技术的优越性。
双向电泳--标准操作(完整版)

细胞裂解(蛋白质提取)一、试剂配置:PBS配方氯化钠(MW 58.44) 130mM 8g氯化钾(MW 74.5) 2.7mM 0.2g十二水和磷酸氢二钠(MW 358) 10 mM3.63g磷酸二氢钾(MW 136) 2 mM0.24g加水至1L配好后进行高压灭菌。
Washing buffer(500mL)配方Tris(MW 121.1) 10mM 0.605g蔗糖(MW 342) 250mM 42.75g加水溶解,用HCl调pH至7.0后用孔径为0.22µm滤膜过滤(使用1M盐酸略高于2750uL调pH)裂解储液配方(细胞):尿素(MW 60.06) 7M 4.2g硫脲(MW 76.12) 2M 1.52gCHAPS(MW614.89) 4%(W/V)0.4g 加超纯水定容至10mL后经0.22µm一次性滤膜过滤,过滤后分装成20小管,每小管500uL,-20℃冰箱保存。
裂解储液配方(组织):尿素(MW 60.06)5M3g硫脲(MW 76.12) 2M1.52gCHAPS(MW614.89) 2% 0.2gTris(MW 121.1)40mM 0.048g加超纯水定容至10mL后经0.22µm一次性滤膜过滤,过滤后分装成20小管,每小管500uL,-20℃冰箱保存。
裂解液:裂解液储液 100uLIPG buffer(pH可选) 2% 2uLPi 2uLNucLease mix(100×) 1uLPMSF(100mM:20mg/mL) 1mM 1uLDTT(0.411g/mL) 40mM 1.5uLPi:每片使用200uL超纯水溶解后按10 uL分装考马斯亮蓝G-250:考马斯亮蓝G-250 0.01% 100g95%乙醇 4.7% 50mlH3PO4 8.5% 85g 将考马斯亮蓝G-250溶于50ml95%乙醇中,与用水溶解的100ml H3PO4混合后稀释至1000ml,之后使用滤纸过滤。
双向电泳法

双向电泳法双向电泳法(Bidimensional Electrophoresis,2-DE)是一种常用的蛋白质分离技术,可以同时分析样品中上千种蛋白质。
本文将详细介绍双向电泳法的原理、步骤和应用。
原理双向电泳法结合了等电聚焦(IEF)和SDS-PAGE两种技术,通过两个维度的分离将复杂的蛋白质混合物分解为一系列单独的斑点。
在第一维度中,根据蛋白质的等电点(pI)进行分离;在第二维度中,根据蛋白质的分子量进行分离。
通过将这两个维度的分离结果叠加,可以获得高分辨率的蛋白质图谱。
双向电泳法的关键步骤如下:1.等电聚焦(IEF):在第一维度中,使用等电聚焦技术将样品中的蛋白质按照其等电点进行分离。
等电聚焦是一种基于蛋白质在电场中向氧化物离子(OH-)或氢离子(H+)方向移动的分离方法。
在等电聚焦过程中,蛋白质会在pH梯度中向其等电点迁移,直到净电荷为零。
通过控制pH梯度和应用的电压,可以将蛋白质在等电聚焦过程中分离开。
2.SDS-PAGE分离:在第二维度中,将第一维度的等电聚焦凝胶与SDS-PAGE凝胶垂直叠加。
在SDS-PAGE凝胶中,蛋白质通过聚丙烯酰胺凝胶的孔隙随着电场的作用向阳极迁移。
由于SDS(十二烷基硫酸钠)的存在,蛋白质在SDS-PAGE凝胶中的迁移速度与其分子量成反比。
因此,蛋白质在SDS-PAGE 凝胶中会根据其分子量进行分离。
3.染色和分析:经过双向电泳分离后,凝胶可以通过染色方法显示出一系列斑点,每个斑点代表一个蛋白质。
常用的染色方法包括银染法、荧光染色、贵金属染色等。
对于银染法,它在灵敏度和线性范围上具有优势。
染色后可以使用成像设备捕捉图像并进行定量分析。
通过对斑点的比较和定量,可以识别不同样品之间的差异和变化。
步骤双向电泳法的步骤如下:1.样品制备:将待分析的生物样品(如细胞提取物)进行蛋白质提取,并使得蛋白质在石蜡中可溶解。
常用的方法包括总蛋白提取、亲和层析、激光捕获等。
2.等电聚焦(IEF):将蛋白质样品与具有连续pH梯度的凝胶进行接触。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
附:双向电泳完整的操作步骤
(一)第一向等电聚焦
1. 从冰箱中取-20℃冷冻保存的水化上样缓冲液(I)(不含DTT,不含Bio-Lyte)一小管(1ml/管),置室温溶解。
2. 在小管中加入0.01g DTT,Bio-Lyte 4-6、5-7各2.5ml,充分混匀。
3. 从小管中取出400ml水化上样缓冲液,加入100ml样品,充分混匀。
4. 从冰箱中取-20℃冷冻保存的IPG预制胶条(17cm pH 4-7),室温中放置10分钟。
5. 沿着聚焦盘或水化盘中槽的边缘至左而右线性加入样品。
在槽两端各1cm左右不要加样,中间的样品液一定要连贯。
注意:不要产生气泡。
否则影响到胶条中蛋白质的分布。
6. 当所有的蛋白质样品都已经加入到聚焦盘或水化盘中后,用镊子轻轻的去除预制IPG胶条上的保护层。
7. 分清胶条的正负极,轻轻地将IPG胶条胶面朝下置于聚焦盘或水化盘中样品溶液上,使得胶条的正极(标有+)对应于聚焦盘的正极。
确保胶条与电极紧密接触。
不要使样品溶液弄到胶条背面的塑料支撑膜上,因为这些溶液不会被胶条吸收。
同样还要注意不使胶条下面的溶液产生气泡。
如果已经产生气泡,用镊子轻轻地提起胶条的一端,上下移动胶条,直到气泡被赶到胶条以外。
8. 在每根胶条上覆盖2-3ml矿物油,防止胶条水化过程中液体的蒸发。
需缓慢的加入矿物油,沿着胶条,使矿物油一滴一滴慢慢加在塑料支撑膜上。
9. 对好正、负极,盖上盖子。
设置等电聚焦程序。
10.聚焦结束的胶条。
立即进行平衡、第二向SDS-PAGE电泳,否则将胶条置于样品水化盘中,-20℃冰箱保存。
(二)第二向SDS-PAGE电泳
1. 配制10%的丙烯酰胺凝胶两块。
配80ml凝胶溶液,每块凝胶40ml,将溶液分别注入玻璃板夹层中,上部留1cm的空间,用MilliQ水(没有milliq的话ddh2o也行,注,水云深浪按)、乙醇或水饱和正丁醇封面,保持胶面平整。
聚合30分钟。
一般凝胶与上方液体分层后,表明凝胶已基本聚合。
2. 待凝胶凝固后,倒去分离胶表面的MilliQ水、乙醇或水饱和正丁醇,用MilliQ水冲洗。
3. 从-20℃冰箱中取出的胶条,先于室温放置10分钟,使其溶解。
4. 配制胶条平衡缓冲液I。
5.在桌上先放置干的厚滤纸,聚焦好的胶条胶面朝上放在干的厚滤纸上。
将另一份厚滤纸用MilliQ水浸湿,挤去多余水分,然后直接置于胶条上,轻轻吸干胶条上的矿物油及多余样品。
这可以减少凝胶染色时出现的纵条纹。
6. 将胶条转移至溶涨盘中,每个槽一根胶条,在有胶条的槽中加入5ml胶条平衡缓冲液I。
将样品水化盘放在水平摇床上缓慢摇晃15分钟。
7. 配制胶条平衡缓冲液II。
8. 第一次平衡结束后,彻底倒掉或吸掉样品水化盘中的胶条平衡缓冲液I。
并用滤纸吸取多余的平衡液(将胶条竖在滤纸上,以免损失蛋白或损坏凝胶表面)。
再加入胶条平衡缓冲液II,继续在水平摇床上缓慢摇晃15分钟。
9. 用滤纸吸去SDS-PAGE聚丙烯酰胺凝胶上方玻璃板间多余的液体。
将处理好的第二向凝胶放在桌面上,长玻璃板在下,短玻璃板朝上,凝胶的顶部对着自己。
10.将琼脂糖封胶液进行加热溶解。
11.将10×电泳缓冲液,用量筒稀释10倍,成1×电泳缓冲液。
赶去缓冲液表面的气泡。
12.第二次平衡结束后,彻底倒掉或吸掉样品水化盘中的胶条平衡缓冲液II。
并用滤纸吸取多余的平衡液(将胶条竖在滤纸上,以免损失蛋白或损坏凝胶表面)。
13.将IPG胶条从样品水化盘中移出,用镊子夹住胶条的一端使胶面完全浸末在1×电泳缓冲液中。
然后将胶条胶面朝上放在凝胶的长玻璃板上。
其余胶条同样操作。
14.将放有胶条的SDS-PAGE凝胶转移到灌胶架上,短玻璃板一面对着自己。
在凝胶的上方加入低熔点琼脂糖封胶液。
15.用镊子、压舌板或是平头的针头,轻轻地将胶条向下推,使之与聚丙烯酰胺凝胶胶面完全接触。
注意不要在胶条下方产生任何气泡。
在用镊子、压舌板或平头针头推胶条时,要注意是推动凝胶背面的支撑膜,不要碰到胶面。
16.放置5分钟,使低熔点琼脂糖封胶液彻底凝固。
17.在低熔点琼脂糖封胶液完全凝固后。
将凝胶转移至电泳槽中。
18.在电泳槽加入电泳缓冲液后,接通电源,起始时用的低电流(5mA/gel/17cm)或低电压,待样品在完全走出IPG胶条,浓缩成一条线后,再加大电流(或电压)(20-30mA/gel/17cm),待溴酚蓝指示剂达到底部边缘时即可停止电泳。
19.电泳结束后,轻轻撬开两层玻璃,取出凝胶,并切角以作记号(戴手套,防止污染胶面)。
20.进行染色。
等电聚焦电泳(IFE,isoelectric focusing electrophoresis)
利用特殊的一种缓冲液(两性电解质)在聚丙烯酰胺凝胶内制造一个pH梯度,电泳时每种蛋白质就将迁移到它的等电点(pI)处,即梯度中的某一pH时,就不再带有净的正或负电荷了。
聚丙烯酰氨凝胶电泳
作用原理
聚丙烯酰胺凝胶电泳是网状结构,具有分子筛效应,它有两种形式,一种是非变性聚丙烯酰胺凝胶,蛋白质在电泳中保持完整的状态,蛋白在其中依三种因素分开:蛋白大小,形状和电荷。
而SDS-PAGE仅根据蛋白分子量亚基的不同而分离蛋白。
这个技术首先是1967年由shapiro建立,他们发现在样品介质和丙烯酰胺凝胶中加入离子去污剂和强还原剂后,蛋白质亚基的电泳迁移率主要取决于亚基分子量的大小,电荷因素可以忽视。
SDS是阴离子去污剂,作为变性剂和助溶试剂,它能断裂分子内和分子间的氢键,使分子去折叠,破坏蛋白分子的二。
三级结构。
而强还原剂如巯基乙醇,二硫苏糖醇能使绊胱氨酸残基间的二硫键断裂。
在样品和凝胶中加入还原剂和SDS后,分子被解聚成多肽链,解聚后的氨基酸侧链和SDS结合成蛋白- SDS胶束,所带的负电荷大大超过了蛋白原有的蛋白量,这样就消除了不同分子间的电荷差异和结构差异。
SDS-PAGE一般采用的是不连续缓冲系统,于连续缓冲系统相比,能够有较高的分辨率。
浓缩胶的作用是有堆积作用,凝胶浓度较小,孔径较大,把较稀的样品加在浓缩胶上,经过大孔径凝胶的迁移作用而被浓缩至一个狭窄的区带。
当样品液和浓缩胶选TRIS/HCL缓冲液,电极液选TRIS/甘氨酸。
电泳开始后,HCL解离成氯离子,甘氨酸解离出少量的甘氨酸根离子。
蛋白质带负电荷,因此一起向正极移动,其中氯离子最快,甘氨酸根离子最慢,蛋白居中。
电泳开始时氯离子泳动率最大,超过蛋白,因此在后面形成低电导区,而电场强度与低电导区成反比,因而产生较高的电场强度,使蛋白和甘氨酸根离子迅速移动,形成以稳定的界面,使蛋白聚集在移动界面附近,浓缩成一中间层。