关于HPLC主成分自身对照法检查有关物质时检测波长确定的讨论
浅议有关物质分析方法验证的接受标准

浅议有关物质分析方法验证的接受标准李正邦(杭州民生药物研究院有限公司,杭州311121)摘要目的:阐述HPLC有关物质分析方法各验证项目的接受标准。
方法与结果:介绍HPLC有关物质分析方法各验证项目的目的和操作,参阅文献并结合实际工作经验,通过对比分析提出其接受标准。
结论:正确理解有关物质分析方法验证的目的,是制定合理接受标准的基础;规范的方法验证需要一个较为公认的接受标准。
关键词:HPLC;有关物质;方法验证;接受标准Discussion on Acceptance Criteria of Analytical Method Validation of Related SubstancesLi Zhengbang (Hangzhou Minsheng Institute for Pharma Research Co., Ltd., Hangzhou 311121 )Abstract Objective: To elaborate the acceptance criteria for each validation subject of HPLC analytical method for related substances. Methods and Results: Introduce the purpose and operation of method validation for HPLC related substances, combining literature research with actual work experience, and propose the acceptance criteria by comparative analysis. Conclusion: Formulating reasonable acceptance criteria is based on understanding correctly the purpose of method validation of related substances. It is necessary to establish more recognized acceptance criteria for standardizing method validation.Key words: HPLC; related substances; method validation; acceptance criteria有关物质主要是指药品在生产过程中带入的起始原料、中间体、反应副产物,以及贮藏过程中的生成降解产物等。
《中华人民共和国药典》利福平及制剂标准中有关物质测定方法的修改建议

·1356·Hemld of Medicine V01.31No.100ctober2012分平均总含量的80%~200%为限度范围,即每克含总转移率不是很高,这可能是制备过程中,麻黄采用水药膏体中含盐酸麻黄碱和盐酸伪麻黄碱两种成分的总煎煮工艺,而麻黄碱和伪麻黄碱均有一定的挥发性,在量应为0.090~0.230 mg。
约100℃因挥发损失而导致转移率偏低。
2010年版《中华人民共和国药典》一部麻黄项下参考文献规定,麻黄含水量不得过9.O%;按干燥品计算,含盐[1] 肖崇厚,杨松松,洪筱坤,等.中药化学[M].上海:上海科学技术出版社,1997.酸麻黄碱(C。
H。
,N0·HCl)和盐酸伪麻黄碱(C,。
H。
,[2] 陈德昌.中药化学对照品工作手册[M].北京:中国医药N0·HCl)的总量不得少于0.80%。
上述测定结果表科技出版社,2000.明,用于中试投料的麻黄饮片符合2010年版《中华人[3] 国家药典委员会.中华人民共和国药典(一部)[M].北民共和国药典》要求。
根据以上测定结果,可以计算3京:中国医药科技出版社,2010:1.批中试样品中盐酸麻黄碱和盐酸伪麻黄碱的平均总转[4] 梁生旺.中药制剂分析[M].北京:中国中医药出版社,移率约46.81%。
2007.3讨论DO I lO.3870/vvdb.2012.10.034 3批中试样品盐酸麻黄碱和盐酸伪麻黄碱的平均《中华人民共和国药典》利福平及制剂标准中有关物质测定方法的修改建议余忠1,朱江2,徐婷婷1(1.湖北省随州市中心医院药剂科,441300;2.湖北省随州市食品药品监督检验所,441300) 摘要目的对2叭0年版《中华人民共和国药典》中利福平及其制剂质量标准中有关物质测定方法存在的问题进行探讨。
方法采用反相高效液相色谱法,Si ncom C。
色谱柱(含碳量为11%)(150m m×4.6mm,5斗m),甲醇一乙腈一0.075mol·L“磷酸二氢钾溶液一1.O mo l·L“枸橼酸溶液(10:50:36:4)为流动相,检测波长为254nm。
制订新药质量标准时有关HPLC法检查有关物质的点滴经验

・经验交流・制订新药质量标准时有关HP LC法检查有关物质的点滴经验Ξ卢彩霞1 林力行21南京金鹰医药科技开发有限公司 (南京 210029) 2江苏吴中苏药医药开发公司 (南京 210009)中图分类号 R927.1 文献标识码 A 文章编号 10072306(2004)03262202 尽管薄层色谱法是“有关物质”检查的首选方法,但由于它的分离能力、定量准确性、重复性等均不及液相色谱法,加上含量测定有时也用液相色谱法,两者可方便地同时进行,故液相色谱法用于检查有关物质就越来越多。
2000年版《中国药典》二部收载液相色谱检查“有关物质”,据作者统计有107个品种,而1990年时仅为7个品种。
“有关物质”是指工艺性杂质,如生产过程中原料、中间体、岐副反应产物和存放过程中产生的降解产物。
用液相色谱法测定“有关物质”的关键是防止微量有关物质峰漏检,除了正确地设定色谱峰检测参数(如斜率、峰宽、最小峰面积等)外,还应注意检测方法。
现版《中国药典》有3种方法:①增加进样量。
即将含量测定时的进样量增加若干倍,以放大增加杂质量,产生较高峰响应信号,使大于噪音水平,以防小峰漏检,如已酸羟孕酮。
②调节测定有关物质检测灵敏度,使高于含量测定时的检测灵敏度,也可使杂质峰较含量测定时扩大,以防漏检,如辅酶Q10。
③在增加进样量的同时,又调节检测灵敏度,如吡喹酮。
这3种方法测定时,主峰高度都已超过记录仪满刻度,超满刻度的程度就是有关物质峰扩大的程度。
可用下式表示:超满刻度%=供试品溶液进样量×对照品溶液调节满刻度%对照品溶液进样量现版药典并未具体规定超满刻度的程度,其存在着不超满刻度(如已烯雌酚)至超满刻度300倍,如吡喹酮。
上述3种做法,第1法最好,又简便,只多配一个浓溶液、多进一次样,可沿用含量测定时条件,不再调试峰参数,更有利的是使原先低于检测限的小峰被检出。
第2法省事省时,用面积归一化法计算时不必另配任何溶液,只是以改变检测灵敏度、扩大小峰响应信号,但若此小峰信号与噪音相近,因此噪音信号也被扩大,小峰仍会漏检(通常这类小峰检出已无意义)。
高效液相色谱波长确定原则

高效液相色谱波长确定原则
高效液相色谱(HPLC)波长确定原则是指在HPLC分析中,通过选择合适的检测波长进行检测,以达到最佳的分离和检测效果。
HPLC分析中,通常会选择比较敏感的检测波长进行分析。
这些波长通常是化合物的最大吸收波长或者是在特定波长下具有特定吸
收峰的波长。
同时,考虑到样品的色谱性质和检测灵敏度,选择合适的波长也是非常关键的。
在HPLC波长确定过程中,常用的方法包括:根据荧光性质选择波长、通过UV-Vis吸收光谱分析选择波长、参照文献中的已知波长等。
值得注意的是,在选择检测波长时,还需要考虑到样品的化学性质和色谱条件。
例如,对于含有多种化合物的混合物样品,需要选择不同的波长进行检测以达到最佳的分离效果;同时,在HPLC色谱条件中,如流速、柱温等参数的控制也会对检测波长的选择产生影响。
综上所述,HPLC波长确定原则需要综合考虑样品的化学性质、色谱性质和检测灵敏度等因素,以选择最合适的检测波长,从而达到最佳的分离和检测效果。
- 1 -。
hplc 主成分自身对照法在药物杂质检查中的原理

hplc 主成分自身对照法在药物杂质检查中的原理下载提示:该文档是本店铺精心编制而成的,希望大家下载后,能够帮助大家解决实际问题。
文档下载后可定制修改,请根据实际需要进行调整和使用,谢谢!本店铺为大家提供各种类型的实用资料,如教育随笔、日记赏析、句子摘抄、古诗大全、经典美文、话题作文、工作总结、词语解析、文案摘录、其他资料等等,想了解不同资料格式和写法,敬请关注!Download tips: This document is carefully compiled by this editor. I hope that after you download it, it can help you solve practical problems. The document can be customized and modified after downloading, please adjust and use it according to actual needs, thank you! In addition, this shop provides you with various types of practical materials, such as educational essays, diary appreciation, sentence excerpts, ancient poems, classic articles, topic composition, work summary, word parsing, copy excerpts, other materials and so on, want to know different data formats and writing methods, please pay attention!在药物杂质检查中,HPLC主成分自身对照法是一种常用的方法,它通过测定药物中的主要成分在HPLC色谱下的保留时间和峰面积,与标准品进行对照比较,以确定药物中杂质的种类和含量。
有关物质测定HPLC方法的建立

有关物质测定hplc方法的建立一、开始之前必须明白:1、有关物质来源的两种途径:1)合成过程中的中间体和副产物;2)储存过程中产生的降解产物;2、分析你的样品结构,熟悉样品的基本性质;3、样品中可能含有的中间体;4、样品的基本稳定性和可能产生的降解产物;二、准备工作1、查阅文献,看看是否同行已经做过类似的工作,有的话就简单了,直接奔主题了。
2、没有文献(苦!),那就很是麻烦了。
1)分析结构,看看有无紫外吸收,有就比较简单,选用紫外检测器就行了,没有的话那就麻烦,可以选用蒸发光散射检测器等;2)分析结构,看看选用何种色谱方式进行分离(正相或者反相色谱);3)分析结构,看看流动相该如何选择,要不要选用一些离子对试剂。
4)分析结构。
5)与同类产品进行比较;三、开工(以紫外检测为例)1、流动相的选择(采用粗品进行选择)1)、有文献,按文献进行优化。
不过我得提醒一下,文献的方法参考是可以的,要想完全按照他的条件做出来是很难的。
2)、没有文献。
a、紫外扫描一下,看看哪里有最大吸收。
将能得到的中间体、副产物、分解产物、样品配成相同浓度,在紫外扫描分光光度计上扫描一下,选择所有物质具有相同的最大吸收处的波长作为测定波长。
要是有pda检测器就不需要这一步了。
b、还是得找一些资料,看看类似的样品的流动相,以它为起始流动相进行选择,如果没有,那就找专业书吧,看看它是怎么教你选择流动相的。
2、最低检测限3、系统适应性试验重复进样5次,记录色谱图,给出系统评价报告4、考察中间体及副产物和样品的分离度。
同时定出中间体在色谱图中的出峰位置,为定质量标准提供依据。
5、考察降解产物和样品的分离情况。
这个一般是通过破坏性试验来考察。
常用的一般是高温、强光、氧化、强酸、强碱五个因素进行考察,通过考察,应该可以知道色谱图中那些峰是由于什么因素产生,这个也可为定质量标准提供依据。
6、根据上面的试验,应该可以知道样品的一些大概情况了,样品中有哪些杂质应该比较明确了。
HPLC法校正因子研究中的几个问题

HPLC法校正因子研究中的几个问题20111207HPLC法具有将不同物质分离后逐一定量的分离分析能力,在药品有关物质检测中发挥着越来越重要的作用,成为药品杂质控制中常用而有效的手段之一。
在杂质对照品法、加校正因子的主成分自身对照法、不加校正因子的主成分自身对照法、峰面积归一化法等几种常用的杂质定量方式中,校正因子的研究对于选择合适定量方式,准确定量杂质具有重要意义,因而成为杂质分析方法研究中的重要内容之一。
但从目前注册申报资料实际情况来看,校正因子的研究和使用中尚存在一些需要进一步思考和关注的问题。
1.校正因子的定义及特点一般来讲,HPLC定量测定中,物质的检测量W与色谱响应值(峰面积等)A之间的比值称为绝对校正因子,即单位响应值(峰面积等)所对应的被测物质的量(浓度或质量);而某物质i与所选定的参照物质s的绝对校正因子之比,即为相对校正因子,即通常所讲的校正因子。
目前校正因子主要用于“加校正因子的主成分自身对照法”定量相关特定杂质,这种定量方式因考虑了杂质与主成分的绝对校正因子的不同所引起的测定误差,将标准物质的赋值信息转化为常数,固化在质量标准中,且不需长期提供标准物质,因而成为现阶段杂质控制较为理想可行的手段。
但这种方法有时会因不同仪器及色谱条件的波动,可产生一定范围的误差,需进行充分的方法耐用性验证,并结合色谱峰定位控制等措施,将误差控制在一定范围内。
2.校正因子的测定在校正因子的研究和使用中,标准物质、色谱条件、溶剂、检测波长等均是重要的影响因素,研究中需要予以关注。
2.1 校正因子的测定需要用到特定杂质及主成分的标准物质,这些标准物质应具备量值准确的特点,符合标准物质(对照品)的相关要求;其次,确定校正因子的分析方法应与最终确定的质量标准方法一致,色谱条件等需经筛选优化后确定,如有变更,需考虑对校正因子的影响,必要时重新确定;第三,要关注影响待测物UV吸收的各种因素,如溶液制备所用溶剂最好与最终确定的流动相相同,检测波长最好在特定杂质及主成分UV曲线的峰或谷处,避开吸收值急剧变化波段,以保证测定方法具有较好的耐用性,并保持测定结果的恒定。
HPLC-加校正因子的主成分自身对照法同时测定奥美沙坦酯氢氯噻嗪片中4种有关物质含量
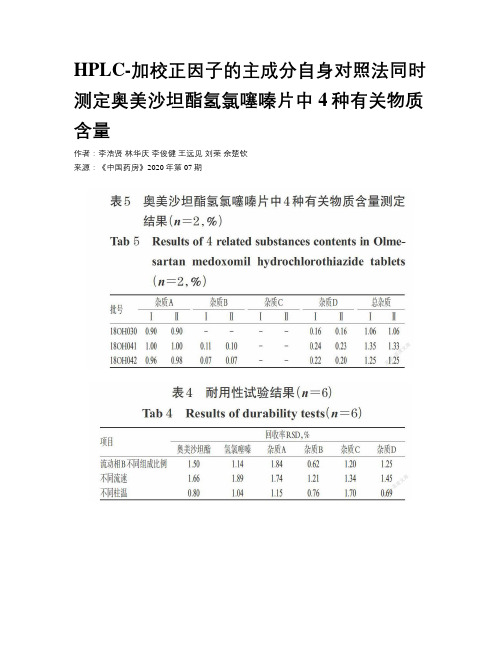
HPLC-加校正因子的主成分自身对照法同时测定奥美沙坦酯氢氯噻嗪片中4种有关物质含量作者:李浩贤林华庆李俊健王远见刘荣余楚钦来源:《中国药房》2020年第07期摘要目的:建立同时测定奥美沙坦酯氢氯噻嗪片中4种已知有关物质(奥美沙坦、奥美沙坦酯二聚体、奥美沙坦酯烯、苯并噻二嗪杂质,简称杂质A、B、C、D)的方法。
方法:采用高效液相色谱(HPLC)-加校正因子的主成分自身对照法进行测定。
色谱柱为YMC-Triart C8;流动相A为乙腈-0.015 mol/L磷酸二氢钾溶液(用磷酸调节pH至2.8)(70 ∶ 30,V/V),流动相B为乙腈-0.015 mol/L磷酸二氢钾溶液(用磷酸调节pH至2.8)(15 ∶ 85,V/V),梯度洗脱;流速为0.8 mL/min;检测波长为265 nm;柱温为25 ℃;自动进样器温度为4 ℃;进样量为10 μL。
结果:杂质A、B、C、D的校正因子分别为1.42、1.17、0.89、0.92。
奥美沙坦酯、氢氯噻嗪和杂质A、B、C、D的质量浓度线性范围分别为0.252 7~7.580 0、1.152 1~4.562 9、0.244 0~18.299 0、0.244 7~3.670 8、0.265 2~3.978 3、 0.149 9~4.497 3 μg/mL(r 均不低于0.999 7),检测限分别为0.084 2、0.050 7、0.081 3、0.081 6、0.088 4、0.050 0μg/mL,定量限分别为0.252 7、0.152 1、0.244 0、0.244 7、0.265 2、0.149 9 μg/mL,中间精密度、稳定性(24 h)、重复性试验结果均符合相关要求,平均回收率分别为104.00%~108.04%、102.00%~104.94%、100.99%~106.89%、92.00%~95.18%、102.00%~105.06%、103.90%~107.00%(n=3)。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
关于HPLC主成分自身对照法检查有关物质时检测波长确定的讨论
审评二部
张玉琥
有关物质检查,包括对产品中残留合成原料、中间体、副产物及可能的降解产物的检查,是控制药品质量的重要指标,目的是检查药品中所含的上述杂质是否符合安全性的要求,同时也是药品稳定性评价中需重点考察的项目。
有关物质检查常用的方法之一是HPLC主成分自身对照法(紫外检测器),即将HPLC色谱图中杂质峰面积与主成分自身对照液峰面积进行比较,以确定杂质限度是否合格。
采用此方法时确定的检测波长是否合理直接影响到方法的可行性,因此检测波长的选择是方法学研究的重要内容。
在审评中发现一些申报单位在采用HPLC主成分自身对照法检查有关物质时直接或间接地以主成分的最大吸收波长作为检测波长,由于有关物质检查的对象是杂质,若将主药的最大吸收波长确定为检测波长,则杂质在此波长下的吸收可能偏低,某些杂质甚至无吸收,这样会造成对杂质含量的低估甚至漏检,从而不能反映产品的真实质量,影响了对品种质量可控性及稳定性的评价。
在有关物质检测波长确定方面,申报资料中比较常见的做法有:
1.直接将主药的最大吸收波长选作检测波长。
2.简单地套用含量测定的色谱条件。
在HPLC法进行含量测定时,为提高方法的灵敏度,降低干扰,往往选用主成分的最大吸收波长作为检测波长。
若套用含量测定的色谱条件,实际仍是以主药的最大吸收波长作为有关物质检测波长。
3.以样品进行破坏性试验(酸、碱、热、光照、氧化等)后的溶液做紫外扫描,将扫描图谱中最大吸收波长确定为有关物质的检测波长。
因破坏性试验后溶液中存在尚未破坏的主药、降解产物、辅料等,此溶液的紫外吸收为各成分紫外吸收的加和,并不能反映降解产物的紫外吸收特性。
由于未破坏主药所占比例较大,故破坏性试验后溶液的最大吸收波长一般仍为主药的最大吸收波长。
采用HPLC主成分自身对照法检查有关物质,其前提之一是需检查的杂质与主成分在确定的检测波长下应有相近的紫外吸收(响应值接近),选择检测波长时需对产品中可能存在的杂质(合成原料、中间体、副产物以及降解产物)的紫外吸收特性进行研究。
已知杂质的紫外吸收特性可采用对其流动相溶液直接进行扫描的方法考察,未知杂质(如未知降解产物等)可通过二极管阵列检测器考察其紫外吸收情况,根据各主要杂质及主成分的紫外吸收特性,选取响应值基本一致的波长作为有关物质的检测波长。
若对不同杂质难于找到均适宜的检测波长,可考虑选择在不同波长下分别测定,也可考虑采用加校正因子的主成分自身对照法。
只有经试验研究确认主成分的最大吸收波长符合有关物质检查对测定波长的要求时,为方便操作,可选作有关物质的检测波长,以与含量测定的色谱条件一致。
另外,HPLC主成分自身对照法检查有关物质比较适用于对微量杂质总量的控制,也可用于单个杂质的限度(一般不超过0.5%)控制。
对于具有明确归属的已知杂质,建议采用杂质对照品法进行检查。
对于有毒有害杂质,更应采用杂质对照品法单独测定,并制定严格的限度。
类别:审评二部。