蛋白印迹分析
蛋白质印迹分析-WesternBlotanalysis

实验四蛋白质印迹分析【实验目的】了解蛋白质印迹法的基本原理及其操作和应用。
【实验原理】蛋白质印迹法又称为免疫印迹法,这是一种可以检测固定在固相载体上蛋白质的免疫化学技术方法。
待测蛋白既可以是粗提物也可以经过一定的分离和纯化, 另外这项技术的应用需要利用待测蛋白的单克隆或多克隆抗体进行识别。
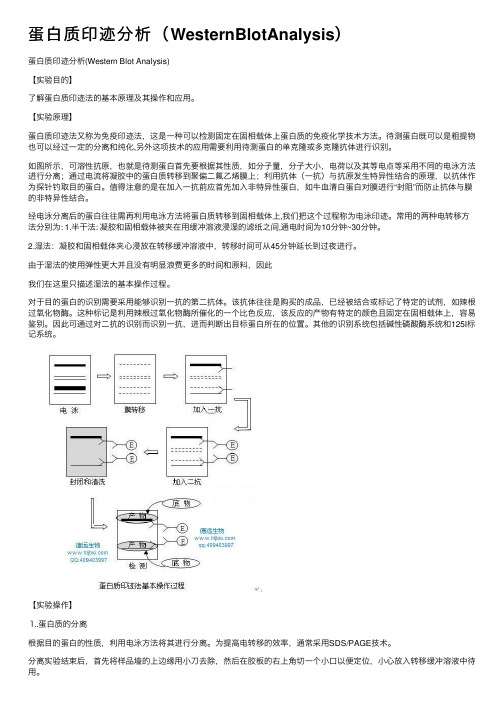
如图所示,可溶性抗原,也就是待测蛋白首先要根据其性质,如分子量,分子大小,电荷以及其等电点等采用不同的电泳方法进行分离;通过电流将凝胶中的蛋白质转移到聚偏二氟乙烯膜上;利用抗体(一抗) 与抗原发生特异性结合的原理,以抗体作为探针钓取目的蛋白。
值得注意的是在加入一抗前应首先加入非特异性蛋白,如牛血清白蛋白对膜进行“封阻” 而防止抗体与膜的非特异性结合。
经电泳分离后的蛋白往往需再利用电泳方法将蛋白质转移到固相载体上, 我们把这个过程称为电泳印迹。
常用的两种电转移方法分别为:1.半干法: 凝胶和固相载体被夹在用缓冲溶液浸湿的滤纸之间,通电时间为10 分钟~30 分钟。
2. 湿法:凝胶和固相载体夹心浸放在转移缓冲溶液中,转移时间可从45 分钟延长到过夜进行。
由于湿法的使用弹性更大并且没有明显浪费更多的时间和原料,因此我们在这里只描述湿法的基本操作过程。
对于目的蛋白的识别需要采用能够识别一抗的第二抗体。
该抗体往往是购买的成品,已经被结合或标记了特定的试剂,如辣根过氧化物酶。
这种标记是利用辣根过氧化物酶所催化的一个比色反应,该反应的产物有特定的颜色且固定在固相载体上,容易鉴别。
因此可通过对二抗的识别而识别一抗,进而判断出目标蛋白所在的位置。
其他的识别系统包括碱性磷酸酶系统和125I 标记系统。
【实验材料】1. 实验器材SDS/PAGE实验相关材料;电转移装置;供电设备;PVDF膜 ( Millipore Immobion-P #IPVH 000 10 ); Whatman 3MM纸;其他工具:镊子、海绵垫、剪子、手套、小塑料或玻璃容器、浅盘。
蛋白质印迹法

化学发光
1、将A和B两种试剂在保鲜膜上等体积混合;1min后,将膜蛋白面朝下与此混合液充分接触;1min后,将膜 移至另一保鲜膜上,去尽残液,包好,放入X-光片夹中。
2、在暗室中,将1×显影液和定影液分别倒入塑料盘中;在红灯下取出X-光片,用切纸刀剪裁适当大小 (比膜的长和宽均需大1cm);打开X-光片夹,把X-光片放在膜上,一旦放上,便不能移动,关上X-光片夹,开 始计时;根据信号的强弱适当调整曝光时间,一般为1min或5min,也可选择不同时间多次压片,以达最佳效果; 曝光完成后,打开X-光片夹,取出X-光片,迅速浸入显影液中显影,待出现明显条带后,即刻终止显影。显影时 间一般为1~2min(20~25℃),温度过低时(低于16℃)需适当延长显影时间;显影结束后,马上把X-光片浸 入定影液中,定影时间一般为5~10min,以胶片透明为止;用自来水冲去残留的定影液后,室温下晾干。
其他信息
其他信息
值得一提的是,Western Blot这个名称的由来很有意思。最开始做印迹工作的是一个叫做Edwin Southern 的科学家,但印迹的对象是DNA链,他把这种技术称为Southern Blot。后来出现了两个过程相似,但是对象不同 的印迹方法,一个针对RNA,一个针对蛋白质,人们把这两种技术分别称为Northern Blot(由斯坦福大学的 George Stark发明)和Western Blot。这两个技术的命名与发明人的名字没有关系了。
原理
原理
与Southern Blot或Northern Blot杂交方法类似,但Western Blot法采用的是聚丙烯酰胺凝胶电泳,被 检测物是蛋白质,“探针”是抗体,“显色”用标记的二抗。经过PAGE(聚丙烯酰胺凝胶电泳)分离的蛋白质样 品,转移到固相载体(例如硝酸纤维素薄膜)上,固相载体以非共价键形式吸附蛋白质,且能保持电泳分离的多 肽类型及其生物学活性不变。以固相载体上的蛋白质或多肽作为抗原,与对应的抗体起免疫反应,再与酶或同位 素标记的第二抗体起反应,经过底物显色或放射自显影以检测电泳分离的特异性目的基因表达的蛋白成分。该技 术也广泛应用于检测蛋白水平的表达。
蛋白质印迹(Western blotting)

蛋白质印迹(Western blotting)一、实验目的:免疫印迹主要用于蛋白质的结构和活性检测等方面,学生掌握与免疫印迹相关的原理和方法,如电泳技术,转膜技术等。
二、实验原理:蛋白质印迹(Western blotting)是将蛋白质转移并固定在化学合成膜的支撑物上,然后以特定的亲和反应、免疫反应或结合反应及显色系统分析此印迹。
蛋白质印迹(免疫印迹)的实验包括5个步骤:1)固定(immobilization):蛋白质进行聚丙烯酰胺凝胶电泳(PAGE),强度比较差,需要从胶上转移到硝酸纤维素膜上。
2)封闭(blocked):保持膜上没有特殊抗体结合的场所,使场所处于饱和状态,用以保护特异性抗体结合到膜上,并与蛋白质反应。
3)初级抗体(第一抗体)是特异性的。
4)第二抗体或配体试剂对于初级抗体是特异性结合并作为指示物。
5)适当保温后的酶标记蛋白质区带,产生可见的、不溶解状态的颜色反应。
三、试剂与器材:试剂:(1)人IgG免疫兔的抗血清;(2)辣根过氧化酶一羊抗兔IgG;(3)PBS缓冲液:NaCl 8 g,KCl 0.2 g,KH2P04 O.24 g,Na2HP04·12H20 2.9 g,加蒸馏水至1000 ml,pH值为7.4;(4)PBS-T缓冲液:PBS缓冲液加O.05 9/6 Tween-20;(5)封闭液:0.5%(质量分数)BSA(用PBS缓冲液配制);(6)底物溶液:A液:溶解30 mg CN在5 ml甲醇中;B液:溶解10 mg DAB在5 ml甲醇中;C液:分别搅拌A液和B液10~15 rain,直到完全溶解,然后将A液和B液混合,加PBS至50 ml,分成10 ml 一份,不用的可冷冻(一20~C)(下次直接化冻运用);D液:取10 ml C液,运用时加10 ml 30%(体积分数)H202;(7)转移缓冲液:0.025 mol/L Tris,O.192 mol/L甘氨酸(glycine),20%(体积分数)甲醇(methan01),pH值为8.3。
免疫印迹分析流程

免疫印迹分析流程
免疫印迹分析(Western Blot)是一种常用的蛋白质分析方法。
它的基本流程包括:
1. 样品制备:提取并定量蛋白质样本。
常用的方法有细胞裂解、组织匀浆等。
2. 电泳分离蛋白:使用SDS-PAGE凝胶电泳分离蛋白质。
根据分子量的不同,蛋白质会在凝胶上分离成不同的条带。
3. 转膜:将凝胶中的蛋白转移到NC膜或PVDF膜上。
4. 滤膜:使用5%脱脂奶粉或BSA在室温封闭1小时,以阻断膜胶的非特异性结合位点。
5. 一抗反应:加入特异性的一抗,孵育过夜。
一抗与目标蛋白特异性结合。
6. 二抗反应:洗涤后加入连接HRP的二抗,孵育1-2小时。
二抗与一抗结合。
7. ECL显色:滴加显影液,HRP催化发光底物氧化产生发光信号。
8. 凝胶成像:曝光、扫描和记录结果。
目标蛋白会出现特异性的条带。
9. 数据分析:通过条带的位置和强度分析目标蛋白的相对分子量和表达量。
以上是免疫印迹分析的基本流程。
根据实验目的,可以选择不同的样品制备方法、凝胶系统、转膜系统等,从而实现对蛋白质的分离、定量和功能分析。
蛋白质印迹分析(WesternBlotAnalysis)
蛋⽩质印迹分析(WesternBlotAnalysis)蛋⽩质印迹分析(Western Blot Analysis)【实验⽬的】了解蛋⽩质印迹法的基本原理及其操作和应⽤。
【实验原理】蛋⽩质印迹法⼜称为免疫印迹法,这是⼀种可以检测固定在固相载体上蛋⽩质的免疫化学技术⽅法。
待测蛋⽩既可以是粗提物也可以经过⼀定的分离和纯化,另外这项技术的应⽤需要利⽤待测蛋⽩的单克隆或多克隆抗体进⾏识别。
如图所⽰,可溶性抗原,也就是待测蛋⽩⾸先要根据其性质,如分⼦量,分⼦⼤⼩,电荷以及其等电点等采⽤不同的电泳⽅法进⾏分离;通过电流将凝胶中的蛋⽩质转移到聚偏⼆氟⼄烯膜上;利⽤抗体(⼀抗)与抗原发⽣特异性结合的原理,以抗体作为探针钓取⽬的蛋⽩。
值得注意的是在加⼊⼀抗前应⾸先加⼊⾮特异性蛋⽩,如⽜⾎清⽩蛋⽩对膜进⾏“封阻”⽽防⽌抗体与膜的⾮特异性结合。
经电泳分离后的蛋⽩往往需再利⽤电泳⽅法将蛋⽩质转移到固相载体上,我们把这个过程称为电泳印迹。
常⽤的两种电转移⽅法分别为: 1.半⼲法: 凝胶和固相载体被夹在⽤缓冲溶液浸湿的滤纸之间,通电时间为10分钟~30分钟。
2.湿法:凝胶和固相载体夹⼼浸放在转移缓冲溶液中,转移时间可从45分钟延长到过夜进⾏。
由于湿法的使⽤弹性更⼤并且没有明显浪费更多的时间和原料,因此我们在这⾥只描述湿法的基本操作过程。
对于⽬的蛋⽩的识别需要采⽤能够识别⼀抗的第⼆抗体。
该抗体往往是购买的成品,已经被结合或标记了特定的试剂,如辣根过氧化物酶。
这种标记是利⽤辣根过氧化物酶所催化的⼀个⽐⾊反应,该反应的产物有特定的颜⾊且固定在固相载体上,容易鉴别。
因此可通过对⼆抗的识别⽽识别⼀抗,进⽽判断出⽬标蛋⽩所在的位置。
其他的识别系统包括碱性磷酸酶系统和125I标记系统。
【实验操作】⒈.蛋⽩质的分离根据⽬的蛋⽩的性质,利⽤电泳⽅法将其进⾏分离。
为提⾼电转移的效率,通常采⽤SDS/PAGE技术。
分离实验结束后,⾸先将样品墙的上边缘⽤⼩⼑去除,然后在胶板的右上⾓切⼀个⼩⼝以便定位,⼩⼼放⼊转移缓冲溶液中待⽤。
蛋白质的印迹分析Westernblotting

一、分子杂交与印迹技术的原理
核酸分子杂交 (nucleic acid hybridization ) 在 DNA 复性过程中,如果把不同 DNA 单链
分子放在同一溶液中,或把 DNA 与 RNA 放在一
起,只要在 DNA 或 RNA 的单链分子之间有一定
的碱基配对关系,就可以在不同的分子之间形
成杂化双链(heteroduplex) 。
(一)DNA印迹技术 (Southern blotting) 用于基因组DNA、重组质粒和噬菌体的 分析。 (二)RNA印迹技术 (Northern blotting) 用于RNA的定性定量分析。 (三)蛋白质的印迹分析 (Western blotting) 用于蛋白质定性定量及相互作用研究。
斑点印迹 Dot blotting
同位素、生物素或荧光染料对它的末端 或全链进行进行标记,称为“探针”然 后用于分子杂交,杂交后通过放射自显 影、荧光检测或显色技术,使杂交区带 显现出来
二、印渍技术的类别及应用
DNA印迹技术
Southern blotting RNA印迹技术 Northern blotting 蛋白质印迹技术 Western blotting 斑点印迹 Dot blotting 原位杂交 in situ hybridization DNA点阵 DNA array DNA芯片技术 DNA chip
Southern Blotting
无水乙醇 离心 酚/氯仿 蛋白酶K SDS
tissue
Paper Towels
(二)RNA印渍技术 Northern blotting
又称为Northern杂交,即RNA-DNA杂交分析。
是一种将RNA从琼脂糖凝胶中转印到硝酸纤维 素膜上的方法。 主要用于检测某一组织或细胞中已知的特异 mRNA的表达水平以及比较不同组织和细胞的 同一基因的表达情况。
蛋白质的Wesrern印迹分析
03
与免疫荧光技术相比, Western印迹分析能够同时检 测多个蛋白质,提供更全面的 蛋白质表达信息。
06 参考文献
参考文献
[请在此处插入参考文献3]
[请在此处插入参考文献2]
[请在此处插入参考文献1]
01
03 02
感谢您的观看
THANKS
02
在实验设计阶段,应充分考虑样 品的代表性,并尽量选择具有代 表性的蛋白质样品进行实验。
选择合适的抗体
根据实验目的和蛋白质目标,选择特异性高、亲和力强的抗体,以确保实验结果的准确性和可靠性。
在选择抗体时,应考虑抗体的来源、特异性和使用浓度等因素,并进行充分的实验验证和质量控制。
避免假阳性结果
确保实验过程中样品的处理和操作规范,避免因操作不当而 产生假阳性结果。
或聚偏二氟乙烯膜。
将膜放入转膜缓冲液中, 使其充分湿润。
在电泳槽中加入转膜缓 冲液,确保膜与凝胶紧
密贴合。
在恒流条件下进行转膜, 使蛋白质从凝胶转移到
膜上。
抗体孵育与显色
洗膜
用洗涤液清洗膜,去除未结合 的一抗。
显色
加入显色剂,如ECL或DAB, 使目的蛋白显色。
一抗孵育
将膜与一抗溶液孵育,使一抗 与目的蛋白结合。
Western印迹分析是一种常用的蛋白 质分析技术,通过将蛋白质从凝胶转 移到膜上,然后与特异性抗体进行反 应,实现对蛋白质的检测和识别。
Western印迹分析的原理
蛋白质样品经过电泳分离后,通过转 膜技术转移到固相支持物(如硝酸纤 维素膜或聚偏氟乙烯膜)上。
通过显色反应(如酶联免疫斑点试 验),检测复合物并获得蛋白质的印 迹图像。
详细描述
在生物学和医学研究中,了解特定蛋白质的表达水平对于理解其生物学功能和疾 病发生发展机制至关重要。通过Western印迹分析,科学家们可以检测蛋白质在 细胞或组织中的表达量,从而评估其在特定生理或病理条件下的活性。
蛋白质免疫印迹定量
蛋白质免疫印迹定量
蛋白质免疫印迹定量分析是一种用于研究蛋白质表达、修饰和互作的技术,可以通过比较条带强度来定量分析蛋白质的表达水平。
以下是蛋白质免疫印迹定量分析的一般步骤:
1. 图像获取:将免疫印迹实验的凝胶图像进行扫描,并转化为数字图像。
2. 条带标准化:通过将目标条带与内参条带进行比较,进行条带强度的标准化。
3. 图像分析:使用图像处理软件进行蛋白质表达水平的进一步分析,如定量测定目标蛋白质在各个样本之间相对变化。
4. 统计学分析:根据图像数据,进行必要的统计学分析,如ANOVA,t 检验等,以确定蛋白质表达的变化是否有显著性。
需要注意的是,蛋白质免疫印迹定量分析的结果会受到实验条件、操作规范、蛋白质表达水平差异等因素的影响,因此在实际操作中需要严格按照实验要求和规范进行,以保证结果的准确性和可靠性。
蛋白印迹实验方法的原理和步骤
蛋白印迹实验,又被称为Western blot,是一种用于检测特定蛋白质的方法。
通过这种实验方法,研究人员可以确定样品中是否含有特定蛋白质,以及其相对浓度。
这一方法在生物医学研究中被广泛应用,对于研究蛋白质结构、功能和相互作用具有重要意义。
本文将介绍蛋白印迹实验的原理和步骤,希望能够帮助读者对这一实验方法有更深入的了解。
一、蛋白印迹实验的原理蛋白印迹实验的原理基于蛋白质的分子量及电荷特性。
当蛋白质被电泳分离后,可以通过膜转移和特异性抗体结合的方式来检测目标蛋白。
其基本步骤包括蛋白电泳分离、膜转移、抗体结合和信号检测。
1. 蛋白电泳分离蛋白印迹实验首先需要对样品中的蛋白进行电泳分离。
这一步骤通过SDS-PAGE或其他电泳方法进行,将蛋白质按照分子量大小进行分离。
分离过程中,蛋白质会在凝胶中形成不同的泳动带,便于后续的检测和分析。
2. 膜转移分离完毕后,需要将凝胶上的蛋白质转移到膜上。
这一步骤通常通过湿式膜转移或半干式膜转移进行,目的是将蛋白质牢固地转移到膜上,并保持其原有的位置关系和相对分子量大小。
3. 抗体结合蛋白转移至膜上后,需要与特异性抗体结合。
这些抗体通常是针对特定蛋白的,并且标记有辅酶或发光物质,便于检测和定量分析。
抗体结合过程需要严格控制条件,确保特异性和灵敏度。
4. 信号检测最后一步是通过染色、荧光或化学发光的方式来检测抗体和蛋白的结合情况。
根据信号的强度和位置,可以确定目标蛋白的存在与否以及其相对浓度。
以上就是蛋白印迹实验的基本原理,下面将介绍蛋白印迹实验的具体步骤。
二、蛋白印迹实验的步骤1. 样品处理首先需要将研究样品进行处理,常见的处理方法包括蛋白溶解、沉淀和浓缩。
处理完毕后,样品中的蛋白质将变得易于电泳分离和检测。
2. 蛋白电泳将处理好的样品加载到SDS-PAGE凝胶上,进行蛋白电泳分离。
这一过程需要严格控制电场强度和时间,以确保蛋白质能够按照分子量大小进行有效分离。
3. 膜转移电泳分离完毕后,需要将凝胶上的蛋白转移到膜上。
蛋白质印记实验流程
蛋白质印记实验流程
蛋白质印记实验是一种常用的生物化学技术,用于研究蛋白质
的结构、功能和相互作用。
下面将介绍一般的蛋白质印记实验流程。
1. 细胞培养和蛋白质提取。
首先,需要培养所需的细胞系,并收集细胞。
然后,用细胞裂
解液裂解细胞膜,释放蛋白质。
接着,通过超速离心将裂解液离心,以获得上清液中的蛋白质。
2. 免疫沉淀。
将待测蛋白质上清液与特异性抗体结合,形成抗原-抗体复合物。
然后,使用蛋白A/G琼脂糖或其他亲和层析介质进行免疫沉淀,将
抗原-抗体复合物沉淀下来。
3. SDS-PAGE电泳。
将免疫沉淀得到的蛋白质样品进行SDS-PAGE凝胶电泳分离。
SDS-PAGE可以根据蛋白质的大小进行分离,从而得到不同的蛋白质
条带。
4. 免疫印迹分析。
将SDS-PAGE凝胶中的蛋白质迁移到聚丙烯膜或硝酸纤维膜上,形成蛋白质印记。
然后,使用特异性抗体结合目标蛋白质,通过化学发光或染色方法检测蛋白质印记。
5. 数据分析。
最后,根据免疫印迹结果,分析蛋白质的表达水平、分子量和相互作用等信息。
通过比较不同样品的免疫印迹结果,可以得出对蛋白质的定量和定性分析。
总结,蛋白质印记实验流程包括细胞培养和蛋白质提取、免疫沉淀、SDS-PAGE电泳、免疫印迹分析和数据分析等步骤。
这一流程可以帮助研究人员深入了解蛋白质的结构和功能,为生物医学研究提供重要的实验数据。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
【实验目的】了解蛋白质印迹法的基本原理及其操作和应用。
【实验原理】蛋白质印迹法又称为免疫印迹法,这是一种可以检测固定在固相载体上蛋白质的免疫化学技术方法。
待测蛋白既可以是粗提物也可以经过一定的分离和纯化,另外这项技术的应用需要利用待测蛋白的单克隆或多克隆抗体进行识别。
如图所示,可溶性抗原,也就是待测蛋白首先要根据其性质,如分子量,分子大小,电荷以及其等电点等采用不同的电泳方法进行分离;通过电流将凝胶中的蛋白质转移到聚偏二氟乙烯膜上;利用抗体(一抗)与抗原发生特异性结合的原理,以抗体作为探针钓取目的蛋白。
值得注意的是在加入一抗前应首先加入非特异性蛋白,如牛血清白蛋白对膜进行“封阻”而防止抗体与膜的非特异性结合。
经电泳分离后的蛋白往往需再利用电泳方法将蛋白质转移到固相载体上,我们把这个过程称为电泳印迹。
常用的两种电转移方法分别为:1.半干法: 凝胶和固相载体被夹在用缓冲溶液浸湿的滤纸之间,通电时间为10分钟~30分钟。
2.湿法:凝胶和固相载体夹心浸放在转移缓冲溶液中,转移时间可从45分钟延长到过夜进行。
由于湿法的使用弹性更大并且没有明显浪费更多的时间和原料,因此我们在这里只描述湿法的基本操作过程。
对于目的蛋白的识别需要采用能够识别一抗的第二抗体。
该抗体往往是购买的成品,已经被结合或标记了特定的试剂,如辣根过氧化物酶。
这种标记是利用辣根过氧化物酶所催化的一个比色反应,该反应的产物有特定的颜色且固定在固相载体上,容易鉴别。
因此可通过对二抗的识别而识别一抗,进而判断出目标蛋白所在的位置。
其他的识别系统包括碱性磷酸酶系统和125I标记系统。
【实验操作】⒈.蛋白质的分离根据目的蛋白的性质,利用电泳方法将其进行分离。
为提高电转移的效率,通常采用SDS/PAGE技术。
分离实验结束后,首先将样品墙的上边缘用小刀去除,然后在胶板的右上角切一个小口以便定位,小心放入转移缓冲溶液中待用。
⒉.电转移⑴准备PVDF膜根据胶的大小剪出一片PVDF膜,膜的大小应略微小于胶的大小。
将膜置于甲醇中浸泡1分钟,再移至转移缓冲溶液中待用。
夹心放置顺序⑵制作胶膜夹心在一浅盘中打开转移盒,将一个预先用转移缓冲溶液浸泡过的海绵垫放在转移盒的黑色筛孔板上,在海绵垫的上方放置经转移缓冲溶液浸湿的3MM纸,小心地将胶板放在3MM纸上,并注意排除气泡。
将PVDF膜放在胶的上方同时注意排除气泡,再在膜的上方放上一张同样用转移缓冲溶液浸湿过的3MM纸并赶出气泡,放置另一张浸泡过的海绵垫,关闭转移盒。
将转移盒按照正确的方向放入转移槽中,转移盒的黑色筛孔板贴近转移槽的黑色端,转移盒的白色筛孔板贴近转移槽的白色端,填满转移缓冲溶液同时防止出现气泡。
⑶电转移连接电源,在4°C条件下维持恒压100v,1小时⒊.免疫检测⑴膜染色断开电源,将转移盒从转移槽中移出,将转移盒的各个部分分开。
用镊子将PVDF膜小心放入一个干净的容器中,用TBS缓冲溶液进行短暂清洗,从膜上剪下一条宽约5mm的膜放入另一个干净的容器中。
将这条膜在染色液中浸泡1分钟,然后在脱色液中脱色30分钟,确定蛋白质已经转移到PVDF膜上。
⑵膜的封闭和清洗对于没有进行染色的膜,首先倒出TBS缓冲溶液,加入3%封闭缓冲溶液,轻轻摇动至少1小时。
倒掉3%封闭缓冲溶液,并用TBS缓冲溶液清洗3次, 每次5分钟。
⑶一抗倒掉TBS缓冲溶液,加入10 ml 0.5%封闭缓冲溶液及适量的一抗,轻轻摇动1小时以上。
从容器中倒出一抗及封闭缓冲溶液,用TTBS缓冲溶液清洗两次,每次10分钟。
⑷二抗倒出TTBS 缓冲溶液,加入5 ml 0.5%封闭缓冲溶液及适量的二抗。
轻轻摇动30分钟,倒出二抗及封闭缓冲溶液,用TTBS缓冲溶液清洗两次,每次10分钟。
⑸检测倒掉TTBS 缓冲溶液,并加入显影剂,轻轻摇动PVDF膜,观察显影情况,当能够清晰的看到显色带时,用蒸馏水在30分钟内分三次清洗PVDF膜以终止显色反应的继续进行。
【实验结果】检查膜上显色结果,蓝紫色带所对应的即是目标蛋白的位置。
【思考题】⒈蛋白质印迹法的特点是什么?⒉请解释什么是BSA?并说明它在本实验中的作用。
⒊请说明二抗在蛋白质印迹法中的生物学功能。
⒋如何保存抗体?Western Blot Analysis【Purpose】Comprehend the theory of Western blotting; understand its basic manipulation and application.【Principle】Western blotting is also called Immunoblotting. It is a kind of immunochemical techniques which is used to detect a protein immobilized on a matrix. The target protein can be in a crude extract or a more purified preparation and the monoclonal or polyclonal antibody against this protein is necessary to help us to recognize the antigen.As in the Figure, soluble antigens (the target protein) may be separated by electrophoresis based on its molecular weight (SDS/PAGE), size and charge (nondenaturating gel electrophoresis or isoelectric point (isoelectric focusing). After the separation, the proteins are transferred from the gel to a PVDF membrane. Once on the membrane antibodies (first antibodies) can be used to probe for the presence of particular protein because of the specifically binding of antigen with against it. Non-specific binding site can be “blocked” using other non-specific protein such as bovine serum albumin before adding first antibody to avoid non-specific binding.Protein transfer is most commonly accomplished by electrophoresis,This procedure is called electrophoretic blotting. The two common electrophoretic methods are:⒈Semi-dry blotting, in which the gel and immobilizing matrix are sandwiched between buffer-wetted filter papers through which a current is applied for 10-30 minutes.⒉Wet (tank) blotting, in which the gel-matrix sandwich is submerged in transfer buffer for electrophoresis, which may take as little as 45 minutes or may be allowed to continue overnightWe only describe wet blotting here, since it permits greater flexibility without being significantly more expensive in time or materials.The detection of target protein is using a second antibody, which can recognize the first antibody. Typically, the second antibody is purchased already conjugated to a labeling agent such as the enzyme horseradish peroxidase. This marker is then visualized by a colorimetric reaction catalyzed by the enzyme which yields a colored product that remains fixed to the membrane. Thus, it is possible to recognize first antibody through recognizing second antibody, and then identify the position of target protein. Other detection systems include alkaline phosphatase and 125I labels.【Materials】⒈Apparatus:Apparatus of SDS-PAGE, Electroblotting Apparatus, Power supply, PVDF membrane (Millipore Immobion-P #IPVH 000 10), Whatman 3MM paper, Additional Tools: Forceps, sponge pad, scissor, gloves, small plastic or glass container, Shallow tray.⒉Reagents:⑴10x transfer buffer (1 L): 30.3 g Trizma base (0.25 M), 144 g Glycine (1.92 M), pH should be 8.3; without adjustment.⑵1x transfer buffer (2 L): 400 ml Methanol, 200 ml 10x transfer buffer, 1400 ml water.⑶TBS buffer: Add 1.22g Tris (10 mM) and 8.78g NaCl(150 mM) to 1L distilled water and adjust pH to 7.5 with HCl.⑷TTBS buffer: 1L TBS buffer add 0.5ml Tween 20 (0.05%).⑸First antibody: antibody against the target protein.⑹Second antibody: goat anti-rabbit-HRP(horseradish peroxidase).⑺3% Blocking buffer (0.5 L): Add 15mg Bovine serum albumin in TBS buffer to final volume 0.5 L, keep at 4°C to prevent bacterial contamination.⑻0.5% Blocking buffer (0.5 L): Add 2.5mg Bovine serum albumin in TBS buffer to final volume0.5 L, keep at 4°C to prevent bacterial contamination.⑼Developing reagent: 1ml chlonoaphthol solution (30mg/ml in methanol), add 10 ml methanol, add TBS buffer to 50 ml and add 30 ul 30% H2O2.⑽Staining buffer: Add 1g amido black 18B (0.1%), 250ml isopropanol (25%) and 100 ml acetic acid (10%) to distilled water with final volume 1L.⑾Destaining buffer: Add 350ml isopropanol (35%) and 2 ml acetic acid(2%) to distilled water with final volume 1L.【Procedure】⒈. Separation of ProteinRun an electrophoretic separation of known antigenic proteins. The method of separation decided by the characters of target protein, but for sufficiently transferring, the most common method is SDS-PAGE.After separation, remove upper side of sample wells with a razor blade. Notching bottom right-hand corner of gel for orientation and put gel in transfer buffer until ready to use. ⒉. Electrotransfer⑴Preparation of membraneCut a piece of PVDF membrane (Millipore Immobion-P #IPVH 000 10) according to the size of gel. Incubate in methanol for about 1 min on a rocker at room temp. Remove methanol and equilibrate membrane in 1x transfer buffer until ready to use.⑵Arrange gel-membrane sandwichIn a shallow tray, open the transfer cassette. Put a well-soaked sponge pad on the black piece of the transfer cassette and a wetted 3MM paper on the sponge pad. Place the gel on the paper and arrange well so that all air bubbles are removed. Lay the PVDF membrane on the top of gel and remove any air bubbles. Place a wetted sheet of 3MM paper over the PVDF membrane and remove the bubble. Covered with the secondwell-soaked pad. Close the sandwich with the white piece of the cassette. Mount the sandwich in the transfer tank; put the black sides near the black side of the device. Fill the buffer tank with the transfer buffer.⑶Electrotransfer:Attach the electrodes. Set the power supply to 100V (constant voltage) for 1h at 4°C.⒊. Immunodetection⑴Membrane stainingDisconnect transfer apparatus, remove transfer cassette, and peel 3MM paper from membrane. Remove the membrane to a small container. Add 10 ml TBS buffer and wash for short time. Cut out one stripe with 5mm width and put in another clean container. Stain this stripe in staining buffer for 1 min. Destain for 30 min in destaining buffer to check whether protein has been transferred from gel to membrane or not.⑵Membrane blocking and washingFor other part of membrane, pour off TBS buffer. Add 3% blocking buffer,rock gently for at least 1 h. Pour off 3% blocking buffer and rinse briefly with TBS buffer three times, 5 minutes for per time.⑶First antibodyPour off TBS buffer. Add first antibody at appropriate dilution in 10 ml 0.5% blocking buffer. Rock gently for at least 1 h; pour off first antibody solution from membrane and wash twicefor 10 minutes with TTBS buffer.⑷Second antibodyPour off TTBS buffer. Add second antibody at appropriate dilution in 5 ml 0.5% blocking buffer. Rock gently for 30 min, pour off second antibody solution from membrane and wash twice for 10 minutes with TTBS buffer.⑸DetectionPour off TTBS buffer from membrane and add developing reagent, Rock PVDF gently, monitoring development. When the bands can be seen clearly, stop development by washing membrane with distilled water for 30 minutes with 3 changes.【Result】Check the bands on membrane, the band with blue-purple color corresponding to the target protein.【Questions】⒈What is the character of western blotting?⒉What is the BSA? And explain its function during this experiment.⒊Please elucidate the function of second antibody during western blotting.⒋How to reserve antibody?。