双向电泳操作步骤
双向电泳详细操作过程
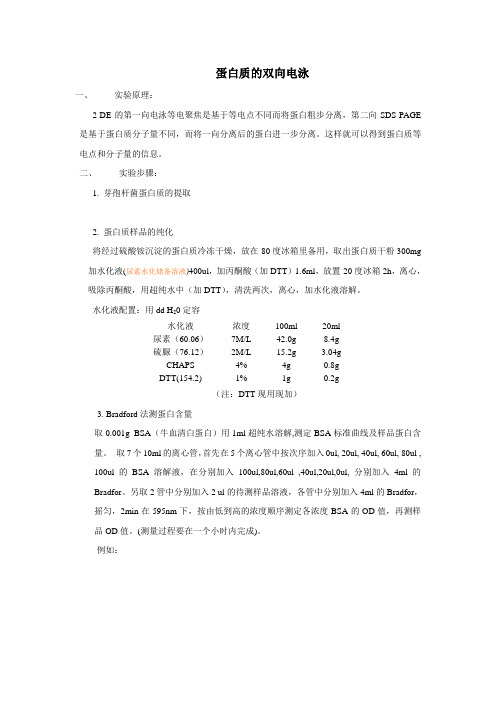
蛋白质的双向电泳一、实验原理:2-DE的第一向电泳等电聚焦是基于等电点不同而将蛋白粗步分离,第二向SDS-PAGE 是基于蛋白质分子量不同,而将一向分离后的蛋白进一步分离。
这样就可以得到蛋白质等电点和分子量的信息。
二、实验步骤:1. 芽孢杆菌蛋白质的提取2. 蛋白质样品的纯化将经过硫酸铵沉淀的蛋白质冷冻干燥,放在-80度冰箱里备用,取出蛋白质干粉300mg 加水化液(尿素水化储备溶液)400ul,加丙酮酸(加DTT)1.6ml,放置-20度冰箱2h,离心,吸除丙酮酸,用超纯水中(加DTT),清洗两次,离心,加水化液溶解。
水化液配置:用dd H20定容水化液浓度100ml 20ml尿素(60.06)7M/L 42.0g 8.4g硫脲(76.12)2M/L 15.2g 3.04gCHAPS 4% 4g 0.8gDTT(154.2) 1% 1g 0.2g(注:DTT现用现加)3. Bradford法测蛋白含量取0.001g BSA(牛血清白蛋白)用1ml超纯水溶解,测定BSA标准曲线及样品蛋白含量。
取7个10ml的离心管,首先在5个离心管中按次序加入0ul, 20ul, 40ul, 60ul, 80ul , 100ul的BSA溶解液,在分别加入100ul,80ul,60ul ,40ul,20ul,0ul, 分别加入4ml的Bradfor。
另取2管中分别加入2 ul的待测样品溶液,各管中分别加入4ml的Bradfor,摇匀,2min在595nm下,按由低到高的浓度顺序测定各浓度BSA的OD值,再测样品OD值。
(测量过程要在一个小时内完成)。
例如:标准曲线方程式:Y= aX+b.其中Y为OD值,X为蛋白含量。
a、b通过作图输入数据可知G250的配置:称取G250 固体0.1g加水定容至1L。
使用前滤纸过滤。
比色皿用70%的乙醇保存,待用时用双蒸水冲洗,再用无水乙醇冲洗,双蒸水冲洗,再加入待测样品溶液润洗,然后,加入样品,测定OD值。
蛋白质双向电泳实验流程

蛋白质双向电泳实验流程一.样品制备1.研磨研磨时间要尽量短,并需及时补充液氮,研磨要充分,同时要保证损失少。
2.重新加入8mltris饱和状态酚(ph8.8)和8ml裂解液,在通风橱内研磨30s。
先加8mltris饱和状态酚,tris饱和状态酚会变为液态,此时需以研磨碓将液态的tris饱和状态酚研磨变成小块。
接着重新加入8ml裂解液,也须要将液态的裂解液研磨变成小块。
等三者搅匀后,将粉末迁移至45mltube。
3.振荡30min。
室温静置,等待tube中液态变为液体后,已经开始震荡。
震荡须要持续30min,每震荡1min,放在冰上加热1min。
4.10000g,4℃,10min。
将酚相(topphase)转移至45mltube。
酚相(topphase)可置于冰上。
酚二者必须就是绿色的,水相必须就是淡黄色的。
5.取6ml的抽提液和6ml饱和酚加入水相,蜗旋振荡30min。
振荡需持续30min,每振荡1min,置于冰上冷却1min。
6.10000g,4℃,10min。
将酚二者(topphase)迁移至45mltube。
7.沉淀酚相。
取一定体积(是酚相的5倍)的0.1m乙酸铵/甲醇溶液(c20℃保存)于酚相(45mltube)。
振荡30s,c20℃培育1h或过夜。
8.冲洗结晶①15min,20,000g,4℃。
弃上清。
②提10ml0.1m乙酸铵/甲醇溶液,用移液器吸打。
c20℃结晶30min。
③15min,20,000g,4℃。
弃上清。
④重新加入10ml乙酸铵/甲醇溶液,用移液器吸打。
c20℃结晶30min。
⑤15min,20,000g,4℃。
弃上清。
⑥提10ml80%丙酮(ice-cold),用移液器吸打。
c20℃结晶30min。
⑦15min,20,000g,4℃。
弃上清。
⑧重新加入10ml80%丙酮(ice-cold),用移液器吸打。
c20℃结晶30min。
⑨15min,20,000g,4℃。
弃上清。
双向凝胶电泳实验方法

双向凝胶电泳实验方法实验方案1.样品制备(Sample preparation)1.1仪器高速离心机1.2试剂NS(制备好的);Citrated human plasma ;溶解缓冲液(9M 尿素,4 , CHAPS,2 , IPG 缓冲液,40 mM DTT,40mM Tris-base) 1.3样品处理方法1).吸取适量的制备好的NS加入到Citrated human plasma 中,在温度为37°下孵化5min。
NS与血浆的加入比例为0.6:8ml。
2)孵化后,将样品高速离心,离心转速为,时间为。
3)除去上清液,将沉淀的纳米球再混悬于0.05M,pH7.4的磷酸缓冲盐中,再次以上次条件离心,本步骤重复三次。
4)将离心后收集到的总蛋白溶解于溶解缓冲液(9M 尿素,4 , CHAPS,2 , IPG 缓冲液,40 mM DTT,40mM Tris-base)。
2.第一相等电聚焦(IEF)2.1固定化 pH 剃度胶条的水化(IPG strip rehydration) 2.1.1仪器IPGphor2.1.2试剂水化液(8 M 尿素,2 % CHAPS,15 mM DTT 和 0.5 % IPG 缓冲液) 水化液需当天新鲜配制2.1.3实验步骤A. 加样品溶涨1).用样品溶解缓冲液(9M 尿素,4 , CHAPS,2 , IPG 缓冲液,40 mM DTT,40mM Tris-base)溶解样品。
蛋白质上样浓度(Q:如何确定上样浓度,)不要超过 10 mg/ml,否则会造成蛋白质的集聚或沉淀。
2).吸取适量(见下表1)含有样品的水化液放入标准型胶条槽中,为确保样品充分进入胶条中,不要加入过量的水化液。
表1 IPG胶条所需水化液体积3).去掉 IPG 胶条的保护膜,胶面朝下,先将 IPG 胶条尖端(阳性端)朝标准型胶条槽的尖端方向放入胶条槽中,慢慢下压胶条,并前后移动,避免生成气泡,最后放下 IPG 胶条平端(阴极),使水化液浸湿整个胶条。
双向电泳操作步骤

双向电泳操作步骤双向电泳操作步骤及相关溶液配置A(实验过程一实验原理:2-DE的第一向电泳等电聚焦是基于等电点不同而将蛋白粗步分离,第二向SDS-PAGE是基于蛋白质分子量不同,而将一向分离后的蛋白进一步分离。
这样就可以得到蛋白质等电点和分子量的信息。
二实验步骤:1. 样品的溶解取纯化后的晶体蛋白3.0mg,加入300ul裂解液(1mg蛋白:100ul裂解液)振荡器上振荡10min左右,共处理一个小时。
其中每隔10,15分钟振荡一次,然后13200rpm离心15min除杂质,取上清分装,每管70ul,—80oC保存。
2. Bradford法测蛋白含量取0.001g BSA(牛血清白蛋白)用1ml超纯水溶解,测定BSA标准曲线及样品蛋白含量。
取7个10ml的离心管,首先在5个离心管中按次序加入0ul, 5ul,10ul, 15ul, 20ul 的BSA溶解液,另2管中分别加入2 ul的待测样品溶液,再在每管中加入相应体积的双蒸水(总体积为80ul),然后,各管中分别加入4ml的Bradford液(原来配好的Bradford液使用前需再取需要的剂量过滤一遍方能使用),摇匀,2min在595nm下,按由低到高的浓度顺序测定各浓度BSA的OD值,再测样品OD值。
(测量过程要在一个小时内完成)。
3. 双向电泳第一向---IEF(双向电泳中一律使用超纯水)3.1 水化液的制备称取2.0mg 的DTT,用700ul水化液储液溶解后,加入8ul 0.05, 的溴酚兰,3.5ul(0.5,v/v)IPG buffer (pH 3-10)振荡混匀,13200rpm离心15min 除杂质,取上清。
在含300ug 蛋白(经验值)的样品溶解液中加入水化液,至终体积为340ul,振荡器上振荡混合,13200rpm离心15min除杂质,取上清。
3.2 点样,上胶分两次吸取样品,每次170ul, 按从正极到负极的顺序加入点样槽两侧,再用镊子拨开 Immobiline DryStrip gels (18cm,pH 3—10)胶条,从正极到负极将胶条压入槽中,胶面接触加入的样品。
双向电泳泳实验方案

双向电泳实验方案1.双向电泳总蛋白(培养细胞)提取方法:方案一细胞培养在10cm的培养皿中,待培养至80%左右密度时,细胞用预冷的PBS漂洗3次,加450μl裂解液,细胞刮刀收集,用移液器转移至1.5ml离心管中,反复吹打。
(折合在六孔板中,每孔加裂解液50μl)之后将样品用超声波细胞破碎仪超声处理超声时间为5s,间歇时间为10s,功率为100-120W,超声处理至溶液清澈无粘稠物为止,处理过程在冰浴中进行,超声处理后,4℃、25000g离心1h。
取上清进行蛋白质浓度测量,按实验所需的量分装后-80℃冰箱中保存。
(裂解液成分:8mol/L脲,65mmol/L DDT,4%CHAPS,40mmol/L Tris)方案二1.1试剂(1)抽提缓冲液9mol/L脲 5.4g4%CHAPS 0.4g0.5%IPG缓冲液(PH 3-10)(AP Biotech) 50.0μl50mmol/L DDT 0.077gH2O 加至10ml(2)IPG缓冲液(PH 3-10)(AP Biotech)(3) 磷酸盐缓冲液(PBS),冰冻1.2仪器(1) 细胞刮刀(2)离心机(低温,低速)(3)滤纸(4)冰浴装置(5)超速离心机(低温)(6)漩涡混合器1.3细胞培养的GC-1 spg细胞1.4方法1.从培养皿中转移细胞:用细胞刮刀从培养皿中刮下细胞,用5ml移液器将培养基和细胞转移至15ml离心管中。
2.480g、4℃离心沉淀细胞5min。
3.弃去上清,勿搅动沉淀要点:操作一下步骤时,所有细胞需保持冰冻状态;不离心或震荡时保持细胞在冰上。
4.离心管中加入10ml冰冻PBS,来回吹打重悬细胞。
5.480g、4℃再次离心细胞5min。
6.弃去上清,勿搅动沉淀。
7.重复步骤4-6两次。
8.在最后一次洗涤后,把离心管完全空干,用滤纸将沉淀上残留的PBS吸干。
9.用移液器将抽提缓冲液加到离心管中。
依赖所研究的细胞系来确定抽提缓冲液的体积。
双向电泳操作步骤

3.1 全菌蛋白的制备将细菌接种于DMEM培养基,置于28℃培养箱培养18h。
参照Coelho 等(Coelho et al. 2004)的方法,略有改动。
用Wash buffer(10mmol/L TrisCl pH8.0,5mmol/L 醋酸镁)清洗细胞3次。
离心,细胞沉淀在Lysis Buffer( 7mol/L 尿素,2mol/L硫尿,1% IPG Buffer pH3-10或pH4-7,4% CHAPS,1% DTT,1%蛋白酶抑制剂,1%核酶抑制剂)中悬浮,使其浓度范围在5~10mg/mL。
置于冰上裂解2h。
13000r/min离心1h取上清,-80℃保存。
用2-D clean-up Kit 纯化蛋白,再用2-D Quant Kit测定样本中蛋白浓度。
3. 2 2-D Clean-Up Kit的使用方法(全程小心)蛋白质样本在1.5mL微型离心管中处理,所有步骤均在冰上进行。
1)将体积100μL的蛋白质样本(含1~100μg蛋白质)置于1.5mL微型离心管中。
加入300μL沉淀剂。
振荡或倒置搅匀。
冰浴中(4~5℃)培育15min。
2)加入300μL共沉淀剂,简单振荡混合一下,12000r/min离心5min。
3)将上清液尽量多地倾析或吸出,不要搅散沉淀。
保持沉淀不变,在其上面加入一层40μL共沉淀剂,冰上培育5min。
后离心5min,去上清。
4)往沉淀加入25μL蒸馏水或去离子水。
将管振荡5~10s,这时沉淀应散开,但并未溶解于水中。
在管中加入1mL洗涤缓冲液(在-20℃下至少预冷1h)和5μL洗涤添加剂,振荡直至沉淀完全散开。
-20℃下培育至少30min,每10min振荡20~30s。
5)将离心管以最大速度(至少12000r/min)离心5min。
小心地将上清液移走弃去。
此时应可见白色沉淀,将沉淀简单风干一下(不要超过5min)。
6)用裂解液溶解沉淀,以备第一向IEF电泳。
振荡管子至少30s,在室温下孵育,振荡或用移液管抽吸使之完全溶解。
蛋白质双向凝胶电泳操作经验及解析

蛋白质双向凝胶电泳操作经验及解析操作步骤:1.样品准备:将待测蛋白质混合物进行样品处理,如蛋白质提取、浓缩和去污等步骤,以获得高纯度、高浓度的样品。
2.第一维电泳:将样品加载到等电点聚焦凝胶中。
等电点聚焦是根据蛋白质的等电点进行分离的方法,即根据蛋白质电荷差异使其定位到等电点位置。
通常使用毛细管等电点聚焦,具体操作步骤是:将样品注入到毛细管中,两端分别连接正负电极,施加电压使得蛋白质开始迁移,直到在等电点位置停止。
这个阶段的电流较低。
3. 第一维凝胶电泳结束后,可使用pH梯度(pH gradient)的凝胶电泳或两种缓冲液浸泡在两边以建立静电场将蛋白质进一步扩散。
4.第二维电泳:将第一维电泳分离得到的凝胶嵌入到另一种凝胶中进行第二次电泳。
通常使用SDS-凝胶。
该凝胶使用离子溶液降解电荷,所有的蛋白质都将带有同样的电荷。
这个阶段的电流较高。
5. 染色和图像分析: 电泳结束后,可以用染色剂进行染色,如Coomassie蓝染色或银染色。
然后使用透射扫描或数字图像分析仪,获取电泳凝胶的图像,并进行质谱分析。
解析与解释:1.蛋白质双向凝胶电泳可以提供更高的分辨率和更好的分离效果,因为它结合了等电点聚焦和SDS-的优势。
2.在等电点聚焦过程中,蛋白质根据其等电点的差异而聚集在凝胶中的不同位置。
这一步骤可以将样品分离成多个窄条带,每个窄条带包含具有相似等电点的蛋白质。
3.在第二维电泳中,蛋白质将根据其分子质量而进一步分离。
较小分子量的蛋白质可以迁移到更远的位置,而较大分子量的蛋白质则停留在较近的位置。
4.通过染色和图像分析,可以将电泳凝胶的图像数字化并用于质谱分析。
这将帮助确定每个蛋白质的分子质量和相对丰度。
蛋白质双向凝胶电泳是一种非常有价值的蛋白质分析方法,尤其适用于分析复杂的混合物。
通过合理的操作步骤和解析方法,可以获得高质量的实验结果。
这些结果对于了解蛋白质的功能和相互作用,以及发现新的生物标志物具有重要的意义。
双向电泳原理及实验步骤

银染(Silver Stain Plus™ stain)
荧光染色(SYPRO® Ruby protein gel stain)
适用于质谱的染色方法
考马斯亮蓝染色
银染的检测灵敏度很高,可达到200pg,但其线性很差。普通的银染过程中因醛类的特异反应,而与下游质谱不兼容。
快速银染法,可与下游质谱兼容,但其检测灵敏度较低,并伴有很深的背景干扰。
聚焦时间的优化
IEF的基本条件
Stemp 1
Stemp 2
Stemp 3
total
voltage
Time
Volt-Hours
Ramp
250
20min
---
Linear
4000
4000
2hr
---
---
10,000V-hr
Linear
Rapid
5 hr
14,000V-hr
7 cm
Stemp 1
Stemp 2
双向电泳样品的溶解
是成功进行双向电泳的最关键因素之一 溶解的目标: 样品中非共价结合的蛋白质复合物和聚积体完全破坏,从而形成各个多肽的溶解液; 必须允许可能干扰2-DE分离的盐、脂类、多糖和核酸等物质的去除; 保证样品在电泳过程中保持溶解状态。
离液剂:通过改变溶液中的氢键结构使蛋白质充分伸展,将其疏水中心完全暴露,降低接近疏水残基的能量域。典型代表是尿素和硫尿。
02
样品上样缓冲液
标准溶液:
Reagent
Amount
8M urea
47ml of 8.5 stock or 24g urea in 25ml H2O
50mM DTT or 2mM TBP
385mg or 500ul of 200mM TBP stock
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
蛋白质的翻译后修饰和加工,是指在肽链合成完成后进行的化学反应,如磷酸化、羟基化、糖基化、二硫键形成,以及目前发现的蛋白质自剪接等等,可能有一百种以上。翻译后修饰和加工对蛋白质的正常生理功能是必需的,它们的变化往往和疾病的发生有关。用双向凝胶电泳可以进行翻译后修饰的研究,如用32P标记可以研究磷酸化蛋白的变化。双向凝胶电泳中常可发现的蛋白质拖曳现象,很可能是一个蛋白的不同翻译后修饰产物所造成的。拖曳图像变化在疾病诊断上可能提供重要的信息。
双向电泳操作步骤
水化上样(被动上样)
1.从冰箱中取出IPG胶条,室温放置10min。
2.沿水化盘槽的边缘从左向右线性加入样品,槽两端各1cm左右不加样,中间的样品液一定要连贯。注意:不要产生气泡,否则会影响胶条中蛋白质的分布。
3.用镊子轻轻撕去IPG胶条上的保护层。注意:碱性端较脆弱,应小心操作。
4.将IPG胶条胶面朝下轻轻置于水化盘中样品溶液上。注意:不要将样品溶液弄到胶条背面,因为这些溶液不会被胶条吸收;还使胶条下面的溶液产生气泡。如产生了气泡,用镊子轻轻地提起胶条的一端,上下移动胶条,直到气泡被赶走。
(4)难溶蛋白的检测,这类蛋白中包括一些重要的膜蛋白。
(5)得到高质量的双向凝胶电泳需要精湛的技术,因此迫切需要自动二维电泳仪的出现。
4存在问题编辑
(1)低拷贝蛋白的鉴定。人体的微量蛋白往往还是重要的调节蛋白。除增加双向凝胶电泳灵敏度的方法外,最有希望的还是把介质辅助的激光解吸/离子化质谱用到PVDF膜上,但当前的技术还不足以检出拷贝数低于1000的蛋白质。
(2)极酸或极碱蛋白的分离。
(3)极大(>200kD)或极小(<10kD=蛋白的分离)。
3.将IPG胶条胶面朝下置于聚焦盘中,胶条的正极(标有+)对应于聚焦盘的正极,确保胶条与电极紧密接触。
4.在每根胶条上覆盖2- 3ml矿物油。
5.对好正、负极,盖上盖子。设置等电聚焦程序。
6.聚焦结束的胶条,立即进行平衡、第二向SDS-PAGE电泳。或将胶条置于样品水化盘中,- 20℃冰箱保存,电泳前取出胶条,室温放置10分钟,使其溶解。
9.将琼脂糖封胶液加热溶解。
10.在100ml量筒中加入TGS电泳缓冲液。
11.第二次平衡结束后,取出胶条,用滤纸吸去多余的平衡液(将胶条竖在滤纸上,以免损失蛋白或损坏凝胶表面)。
12.用镊子夹住胶条的一端使胶面完全浸末在1×电泳缓冲液中漂洗数次。
13.将胶条背面朝向玻璃板,轻轻放在长玻板上,加入低熔点琼脂糖封胶液。
5.将胶条转移至样品水化盘中,加入6ml(17cmIPG)平衡缓冲液I,在水平摇床上缓慢摇晃15分钟。
6.配制胶条平衡缓冲液II。
7.第一次平衡结束后,取出胶条将之竖在滤纸上沥去多余的液体,放入平衡缓冲液II中,继续在水平摇床上缓慢摇晃15分钟。
8.用滤纸吸去SDS-PAGE胶上方玻璃板间多余的液体,将二向凝胶放在桌面上,凝胶的顶部面对自己。
2、样品浓度大于2 μg/ μl;
样品制备
双向电泳成功的关键在于建立一套有效的、可重复的样品制备方法。样品制备的影响因素包括蛋白质的溶解性、分子量、电荷数及等电点等。对于不同的样品性质及研究目的,其方法也不尽相同。不同的样品性质,用于样品处理的缓冲液必须在低离子强度的基础上,既能保持蛋白质的天然电荷。又能维持其溶解性。因此,绝大多数样品往往都需要通过多次实验才能摸索到最适宜的条件。
5.放置30~45min大部分样品被胶条吸收,沿着胶条缓慢加入矿物油,每根胶条约3ml(17cmIPG),防止胶条水化过程中液体蒸发。
6.置等电聚焦仪于- 20℃水化11~15h。
第一向等电聚焦
1.将纸电极置于聚焦盘的正负极上,加ddH2O 5~8μl润湿。
2.取出水化好的胶条,提起一端将矿物油沥干,胶面朝下,将其置于刚好润湿的滤纸片上杂交以去除表面上的不溶物。
第一向分离
等电聚焦
蛋白质是两性分子,在不同的pH环境中可以带正电荷、负电荷或不带电荷。对每个蛋白质来说都有一个特定的pH,此时蛋白质的静电荷为零,此pH值即该蛋白质的等电点(pI)。将蛋白质样品加载至pH梯度介质上进行电泳时,它会向与其所带电荷相反的电极方向移动。在移动过程中,蛋白分子可能获得或失去质子,并且随着移动的进行,该蛋白所带的电荷数和迁移速度下降。当蛋白质迁移至其等电点pH位置时,其净电荷数为零,在电场中不再移动。聚焦是一个与pH相关的平衡过程:蛋白质以不同的速率靠近并最终停留在它们各自的pI值;在等电聚焦过程中,蛋白质可以从各个方向移动到它的恒定位点。
第二向SDS-PAGE电泳
1.配制12%的丙烯酰胺凝胶。
2.待凝胶凝固后,倒去分离胶表面的MilliQ水、乙醇或水饱和正丁醇,用MilliQ水冲洗。
3.配制胶条平衡缓冲液I
4.在桌上先放置干的厚滤纸,聚焦好的胶条胶面朝上放在干的厚滤纸上。将另一份厚滤纸用MilliQ水浸湿,挤去多余水分,然后直接置于胶条上,轻轻吸干胶条上的矿物油及多余样品,这样可以减少凝胶染色法找到感兴趣的蛋白后.就可以从凝胶中或膜上切取这些目标蛋白质作鉴定。现在绝大多数蛋白质的鉴定是通过质谱分析来完成的。
分离蛋白质组所有蛋白
分离蛋白质组所有蛋白的两个关键参数是其分辨率和可重复性。在目前情况下,双向凝胶电泳的一块胶板(16cm×20cm)可分出3~4千个,甚至1万个可检测的蛋白斑点,这与10万个基因可表达的蛋白数目相比还是太少了。80年代开始采用固定化pH梯度胶,克服了载体两性电解质阴极漂移等许多缺点而得以建立非常稳定的可以随意精确设定的pH梯度。由于可以建立很窄的pH范围(如0.05U/cm),对特别感兴趣的区域可在较窄的pH范围内做第二轮分析,从而大大提高了分辨率。此种胶条已有商品生产,因此基本上解决了双向凝胶电泳重复性的问题。这是双向凝胶电泳技术上的一个非常重要的突破。第二向SDS-PAGE有垂直板电泳和水平超薄胶电泳两种做法,可分离10~100kD分子量的蛋白质。
第二向分离
SDS.聚丙烯酰胺凝胶电泳
双向电泳的第二向是将IPG胶条中经过第一向分离的蛋白转移到第二向SDS.PAGE凝胶上,根据蛋白相对分子质量或分子量(MW)大小与第一相垂直的分离。
蛋白质与十二烷基硫酸钠(SDS)结合形成带负电荷的蛋白质.SDS复合物,由于SDS是一种强阴离子去垢剂.所带的负电荷远远超过蛋白质分子原有的电荷量,能消除不同分子之间原有的电荷差异,从而使得凝胶中电泳迁移率不再受蛋白质原有电荷的影响.而主要取决于蛋白质分子的质量大小,其迁移率与分子量的对数呈线性关系,在蛋白质组研究中。需要在同样的条件下同时走多块胶,这对凝胶与凝胶之间的比较十分重要。
其中灵敏度较高的银染色法可检测到4ng蛋白,最灵敏的还是用同位素标记,20ppm的标记蛋白就可通过其荧光或磷光的强度而测定。用图像扫描仪、莱赛密度仪、电荷组合装置可把用上述方法得到的蛋白图谱数字化,再经过计算机处理,去除纵向和横向的曳尾以及背景底色,就可以给出所有蛋白斑点的准确位置和强度,得到布满蛋白斑点的图像,即所谓“参考胶图谱”。蛋白质组研究的主要困难是对用双向凝胶电泳分离出来的蛋白,进行定性和定量的分析。最常用的方法是先把胶上的蛋白印迹到PVDF(polyvinylidene difluoride)膜上后再进行分析,确定它们是已知还是未知蛋白。现在的分级分析法是先做快速的氨基酸组成分析,也可先做4~5个循环的N末端微量测序,再做氨基酸组成分析;结合在电泳胶板上估计的等点电和分子量,查对数据库中已知蛋白的数据,即可作出判断。有文献报道,N末端4个残基序列的数据就可以给出很多的信息而得到相当准确的结果。如再结合C末端序列,判断结果的准确性会更高。对分离得到的蛋白质作进一步的确切鉴定需要有足够数量的纯蛋白,电泳时蛋白质已经过了高度纯化。现在一块胶板可允许上到高达mg数量级的样品,因此每个分离的蛋白斑点可有μg数量的蛋白,这样使本来是微量的蛋白也可希冀被鉴定。
原理编辑
1、根据蛋白质的等电点(第一向)和分子量(第二向)的不同进行分离。
2、电泳后根据蛋白质的上样量对胶进行考马斯亮兰染色、银染或荧光染色,然后用相关软件对电泳图象进行分析。
操作步骤编辑
样品要求
1、建议使用的蛋白质溶解体系为8M尿素/4%CHAPS /40mMTris(Base)/65mM DTT;
3技术应用编辑
凝胶中蛋白的检测
凝胶染色的目的是使其中的蛋白质能够被观察到。目前还没有通用的染色方法,只能在考虑多种因素如需要的灵敏度、线性范围、方便程度、费用、以及成像设备类型等基础上,结合实际进行选择。有时也可以将蛋白转膜后通过免疫印迹的方法来进行检测。
图像采集和分析
成像设备可以摄人图像,对凝胶图像以数字形式保存,对每块凝胶图像进行平等的比较,在各研究组之间传递信息,并对大量的数据进行归类分析。目前应用较为广泛的图像分析软件有PDQuest、ImageMaster 2D Elite、Melanie、BioImage Investigator等,分辨率较高,功能齐全。尽管如此,仍不可避免约10%的未检出点和假点,需要手工添加、删除和分割。图像采集完成后。可以应用各种分析软件进行分析,可以收集、诠释、比较蛋白质组数据资料等。
14.用适当厚度的胶片,轻轻地将胶条向下推,使之与聚丙烯酰胺凝胶胶面完全接触。注意:不要在胶条下方产生气泡,应推动凝胶背面的支撑膜,不要碰到面胶。
15.放置5分钟,使低熔点琼脂糖封胶液凝固。
16.打开二向电泳制冷仪,调温度为15℃。
17.将凝胶转移至电泳槽中,加入电泳缓冲液,接通电源,起始时用的低电流(5mA~10mA/gel/17cm),待样品在完全走出IPG胶条,浓缩成一条线后,再加大电流(20-30mA/gel/17cm)待溴酚蓝指示剂达到底部边缘时即可停止电泳。
18.电泳结束后,轻轻撬开两层玻璃,取出凝胶,并切角以作记号(戴手套,防止污染胶面)。
19.进行染色。
双向凝胶电泳的原理是第一向基于蛋白质的等电点不同用等电聚焦分离,第二向则按分子量的不同用SDS-PAGE分离,把复杂蛋白混合物中的蛋白质在二维平面上分开。近年来经过多方面改进已成为研究蛋白质组的最有使用价值的核心方法。