不对称双羟基化反应
不对称双羟化反应合成

不对称双羟化反应合成(2S )-普萘洛尔盐酸普萘洛尔是白色或乳白色结晶性粉末,无臭,味微甜后苦,熔点 96℃,。
熔点 163~164o C ,干燥失重≤0.5%。
在有机溶剂中溶解能力有较大差异,以甲醇中溶解量最大。
水溶液pH 值为5.0~6.0,遇光着色。
它是一种具有稳定细胞膜作用、无内源性拟交感活性的β受体阻滞剂,临床上用于治疗高血压、心绞痛和快速性心律失常等病症的药物,疗效良好。
通过查阅文献发现,第一篇有关普萘洛尔的文献发表于1965年,在这之后,合成盐酸普萘洛尔的文献陆续见诸报道,其中基本都是英文文献,国内关于合成的报道很少,并且很大一部分为专利,最早的发明专利始见于 1976年。
对其合成方法的研究一直是化学工作者们的科研热点。
文献报道的合成方法主要有两类: 一类是先合成外消旋体,然后拆分;另一类是直接合成(S)- 异构体。
1997年,赵军等人[1] 以(R)-1,2-丙酮缩甘油为原料,经过甲基苯磺酰氯酯化、1-萘酚亲核取代、环状亚硫酸酯化及异丙胺基化等5步反应,合成(S)-普萘洛尔(图1),总收率仅41%,且步骤烦杂,工业化生产比较困难。
2003,朱长生[2]报道了以1-萘酚为原料与环氧氯丙烷反应得1-萘基环氧丙基醚,再与异丙胺作用得外消旋普萘洛尔,最后经L -(+)-酒石酸拆分得(S )-和(R )-普萘洛尔。
该工艺路线中的拆分剂L -(+)-酒石酸比较昂贵, 故生产成本较高。
2007年,王燕等人[3]报道了一种新的普萘洛尔异构体的不对称合成方法, 先以手性Salen-CoIII 即[(R,R)-N,N'-双-(3,5-二叔丁基水杨基)-1,2-环己二胺(2-)]乙酸钴催化剂水解动力学拆分外消旋环氧氯丙烷得到高光学纯度的(S)-环氧氯丙烷和(R)-3-氯-1,2-丙二醇。
以(S)-环氧氯丙烷为手性原料先水解得(S)-3-氯-1,2-丙二图1以(R )-1,2-丙酮缩甘油为原料合成(S )-普萘洛尔醇, 其与1-萘酚反应得(S)-3-(1-萘基)-丙烷-1,2-二醇, 再与氯化亚砜反应得环状亚硫酸酯, 最后和异丙胺作用得(S)-普萘洛尔(图2),总收率80.9%,光学纯度大于99%,该方法合成步骤繁琐,但反应副产物少, 收率高,且光学纯度高。
用不对称二羟基化反应合成(S)-美托洛尔

Abta t A y 4 ( 一 toy ty) h nle e ( )w spe ae rm 4 ( - e oyty) hn l s c : U l 一2me xe 1 p e y t r 2 a rp rdf 一 2m t x e 1 p eo r h h h o h h
aday b md e r ec 2O .( ) -4( - e oyt 1p eoy 一,- oaei n l r i i t e ne f C 3 S - [・2m t xe y)hnx]12p pnd l ll o e n h p s oK 3 h h r o ( 8 %e)w ¥ b ie yay me c i doyao f i erai ces lbsc coa 3, 6 e a otndb sm t h rxli o2wt t dlacsie i i hn a i r dy tn h h e y b —n a a i dd av ,-i 90q i d e pt a n s e h a lad 3cne e t S 一4 l ld e vf e 4b (- 一u in ) h a z e i n . ov ̄ dio( )[一 ko i 1 s ni h i at c r i l h l g n ( -e oyty) hnx ] e y x i e 4 yoept to .( )me po lnoea ido 2m t xe 1peoy m t l ia ( )b n-o h d S 一 t rl vrl e f h h h o rn me o oi ly l
S nh s f( 一 tp oo yA y y tei o )meo rll smmercDiy r x lt n s b ti h d o yai o
XI ANG h n S u , KUANG n . m Yo g q g , DENG o 1n , Ba .i g
锇催化烯烃双羟基化的反应原理与立体选择性

锇催化烯烃双羟基化的反应原理与立体选择性摘要介绍了四氧化锇催化不对称烯烃双羟基化的反应原理和以 N 甲基吗啉 N 氧化物( N methylmorpholine N oxide,NMO)、铁氰化钾K3[Fe(CN) 6]为氧化剂时烯烃双羟基化反应的催化循环;此外,还介绍了该催化反应立体选择性的机理和一些研究进展以及催化体系中手性配体的选择。
关键词四氧化锇烯烃双羟基化反应立体选择性四氧化锇(OsO4)催化的烯烃双羟基化反应是烯烃的一个重要反应,它在有机和天然产物的合成中有着广泛的应用[1,2]。
例如,利用烯烃双羟基化反应可以方便地制备重要的精细化工原料和有机合成中间体——邻二醇,该反应也可以应用于绿色高分子材料的合成等[3]。
OsO4催化烯烃双羟基化反应的一个非常有趣的特点是在合适配体的存在下将表现出很高的立体选择性。
1980年,Sharpless发现手性配体可以诱导该反应的立体选择性[4],从而开创了烯烃不对称双羟基化反应的先河。
之后,Sharpless的研究小组又发现了具有高立体选择性的配体金鸡纳碱衍生物[5],进一步完善了烯烃不对称双羟基化反应,Sharpless 因在这方面的突出贡献荣获了2001年的诺贝尔化学奖[6]。
烯烃的不对称双羟基化反应已成为现代有机合成化学中最重要的反应之一,相关课题仍然是当今化学研究的一个活跃领域[7,8]。
本文介绍OsO4催化烯烃双羟基化的反应原理和立体选择性。
1 化学反应原理1.1 反应历程烯烃双羟基化反应使用的催化剂通常有锇、钌和锰的氧化物,其中OsO4对烯烃加成的选择性和可靠性最高[9]。
OsO4催化烯烃双羟基化反应的反应过程可以用图1表示。
在没有配体存在时,OsO4与烯烃加成应形成锇酸酯,当反应体系有三级胺配体存在时,则形成锇酸酯与胺的配合物;然后,这些锇酸酯中间产物经过氧化或还原水解后即生成邻二醇。
还原水解使用的还原剂有亚硫酸钠、四氢铝锂、硫化氢等,锇酸酯经还原后其中的锇(VI)变成金属锇析出,所以还原水解时,OsO4与烯烃的反应是化学计量的,即生成1摩尔产物需要消耗1摩尔的OsO4。
sharpless不对称双羟基化反应

sharpless不对称双羟基化反应谢尔浦斯不对称双羟基化(Sharpless asymmetric dihydroxylation)反应首次由哈勃·谢尔浦·斯(K. Barry Sharpless)在2001年提出,被公认为中和不对称催化的一大突破,并在2001年荣获诺贝尔化学奖。
谢尔浦斯不对称双羟基化反应以马来酰胺为催化剂,将具有官能团双烯烃氧化成具有手性的双羟基官能团双烯烃,是生物合成中合成多种手性有机分子和抗生素的催化有机反应。
谢尔浦斯不对称双羟基化反应包括加氧剂选择性氧化反应、通过促进氧化反应而进行不对称羟基化,催化药物氧化进行可调节的不对称羟基化反应,以及还原反应实现手性高选择性的不对称羟基化反应等。
早期的谢尔浦斯不对称双羟基化反应的研究着重于马来酰胺的制备工艺及其究竟是何种形态的问题,但是由于催化剂具有较强的质子供体能力,使得马来酰胺催化剂不受酸或碱条件变化的影响,发展成为催化不对称有机氧化反应的理想结构。
如今,谢尔浦斯不对称双羟基化反应已经成为生物合成中不可或缺的重要反应,在医药合成以及金属有机化学领域也有着广泛的应用。
近年来,谢尔浦斯不对称双羟基化反应的研究也取得了突破性进展,包括新型的谢尔浦斯不对称双羟基化反应、底物诱导的不对称羟基化反应等。
谢尔浦斯不对称双羟基化反应在不断被科研人员发掘其实质之后,也极大地促进了药物合成及其它有机反应的不对称催化机理的发展。
谢尔浦斯不对称双羟基化反应是一类较新的现代不对称催化有机反应,拥有容易操作、高效率及高选择性等优点,已成为一项科学上普遍重视的研究领域。
以谢尔浦斯不对称双羟基化反应为基础的合成策略不仅提供了新的合成选择,而且可以迅速为对特定有机分子的合成提供解决方案,从而发挥出谢尔浦斯不对称双羟基化反应在药物合成和药物研发以及其他临床应用中的重要作用。
几类重要的不对称反应及新型手性配体

几类重要的不对称反应及新型手性配体欧阳志强1 欧阳迎春2(1.南昌大学材料科学与工程学院 南昌330047 2.江西师范大学理电学院南昌330027)摘 要:不对称合成是有机合成领域的热点,本文综述了以有机配体———金属配合物为手性催化剂的不对称合成的最新进展。
关键词:不对称合成 手性催化剂 手性配体 引 言自1968年美国孟山都公司的K nowlex和德国的H omer 分别发表了手性膦配体与铑配合物组成的手性催化剂进行的不均相催化氢化以来,人们相继研究开发了一大批具有立体选择性和高催化活性的新型手性配体,本文将就这方面的最新进展作一综述。
1 C =C 双键的不对称氢化反应2,2′一二(二苯基磷)一1,1′—联萘(BI NAP )的Ru络合物还年广泛用于C =C 双缝的不对称氢化。
主要有1,1′一二取代的不含杂原子的烯的不对称氢化;α,β—不饱和和β,γ—不饱和酸的不对称氢化———这类底物的不对称氢化应用于非麻醉性消炎药萘普森和异丁基布洛芬的工业生产上,潜力极大;以及前手性烯丙基醇的不对称氢化:产物为(R )一或(S )一香茅醇,ee 值高达96%-99%,香茅醇是合成L —薄荷醇的中间体。
结构与BI NAP 相似的2,2′—二氨基—1,1′一联萘(BI 2NAM )或其衍生物配体的Rh 配合物可以以36%-95%的光学收率催化a 一酰胺基丙烯酸与H 2的加成,以及前手性烯酰胺的不对称氢化,前手性酮的不对称还原。
Perea 等对平面手性二茂铁的双膦配体、氮膦配体、硫膦配体与Rh ,Ir 或Ru 形成的催化剂对C =C 的不对称氢化进行了考察,指出Rh 催化剂效果最好。
并对其反应底物的结构,溶剂效应及反应动力学等方面进行研究。
2 C =0双键的不对称还原加氢反应。
前手性酮的不对称还原得到光学活性的仲醇,2,2′—二羟基一1,1—联萘(BI NO L )改造的LiAIH 4还原剂(BI NO L -H )用于前手性不饱和酮的还原可得100%ee的相应仲醇,立体选择性依赖于温度、底物、溶剂、配位体等。
手性药物的不对称催化合成

• 1.3生物催化的水解反应
• 生物催化水解反应就是利用生物酶或者微生物催化外消旋化合物中两
个对映体水解或酯交换反应的速度不同,而拆分获得两个光学活性产 物。目前,利用灰色链霉菌蛋白酶和枯草杆菌蛋白酶对氨基酸酯的选择 性水解,拆分合成广谱抗生素氯霉素和Florfenicol 所需中间体,已取得 开创性进展。 生物催化法反应条件温和易于控制,有高度的立体选择性,生成的产 物单一,副产物较少,并且回收率高,无污染。还有一个优点就是可 以完成一些合成难度较高的反应,在手性药物的合成中的应用十分广 泛。
不对称催化合成的定义和分类不对称催化合成方法catalyticchiralreaction使用手性催化剂来控制不对称合成在非手性底物进行不对称反应时加入少量的手性催化剂使它与反应底物或试剂形成高反应活性的中间体催化剂作为手性模板控制反应物对映面经不对称反应得到新的手性产物而手性催化剂在反应中循环使用达到手性增值chiralitincrement或手性放大效应chiralityamplification的效果
• S-萘普生( Naproxen)是80 年代末推出的一种非甾体高效解热镇痛药
图(1)不对称催化合成萘普森新工艺
图(2)不对称催化合成薄荷醇新工艺
• 2.2不对称催化氧化反应
• 目前使用的不对称催化反应主要有两种。一种是环氧化反应,其中烯
丙醇的Sharpless 环氧化反应最为经典,Sharpless 环氧化反应具有简 易性,可靠性,光学纯度高,产物的绝对构型可以预见等优点。它利用钛 试剂作为催化试剂参与烯丙醇的环氧化,是目前为止最成功的环氧化方 法。其通式如下:
• 2.4不对称催化环丙烷化
手性环丙烷结构广泛地存在于天然和人工合成的产物中,例如下述化 合物。日本住友公司用一定摩尔分数的手性铜催化剂催化烯烃发生不 对称环丙烷化反应,合成了二肽抑制剂cilastatin
双羟基化反应

双羟基化反应
Upjohn双羟基化反应(Upjohn Dihydro某ylation)
Upjohn双羟基化反应,是指烯烃在四氧化锇作为催化剂、NMO作再氧化剂的条件下,转化为相应的顺式邻二醇。
此反应避免了使用有毒且昂贵的计量锇试剂。
反应缺点是所需反应时间有时较长,邻二醇在反应条件下易被继续氧化为邻二酮,以及产率较类似的Sharpless不对称双羟基化反应为低。
反应机理
四氧化锇与烯烃发生[3+2]加成、生成锇酸酯中间体,然后转化为相应的顺式邻二醇。
使催化剂再生,必须通过加水分解锇酸酯。
为此,反应通常在含水体系中进行。
加水分解是该催化体系的限速步骤。
不对称羟醛缩合aldol反应
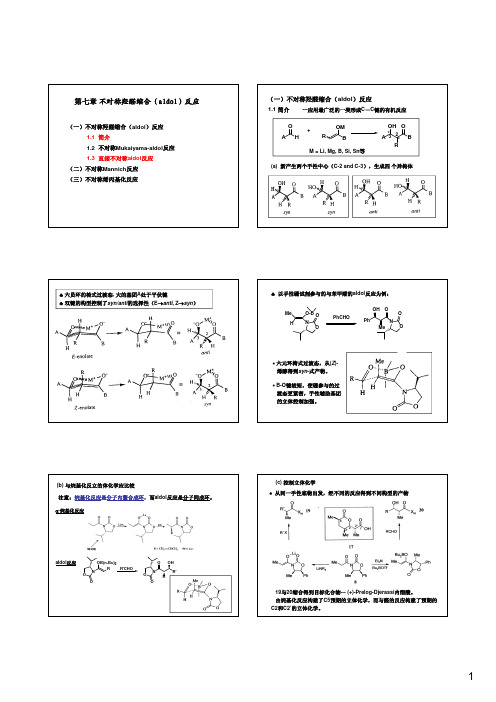
♣以手性硼试剂参与的与苯甲醛的aldol 反应为例:Me O-B NOHPhCHONOPhO Me OHO O ∙六元环椅式过渡态,从(Z )-(b)与烷基化反立的体化学应比较注意:烷基化反应是分子内螯合成环,而aldol 反应是分子间成环。
α-烷基化反应ON R OB(n-Bu)2OR'CHOONO OR'R OH aldol 反应∙从同一手性底物出发,经不同的反应得到不同构型的产物19与20缩合得到目标化合物---(+)-Prelog-Djerassi 内酯酸。
由烷基化反应构建了C5预期的立体化学,而与醛的反应构建了预期的C2和C2’的立体化学。
选择性。
Lewis 酸参与,环状中间体与醛反应,给出syn 构型产物。
Lewis 酸,形成开链(非环)结构,给出anti 构型产物。
醛羰基更易与Lewis 酸配位两类羟醛缩合试剂分别控制syn 和anti 选择性从4得到anti -aldol 产物,而从5得到syn -aldol 产物(2) Mukaiyama-aldol 反应R 1HO +R2R 3OSiMe 31)Lewis acid or R 1R 3OH R 2O Lewis base R 1R3OH R 2O+syn -anti -2)H 2O(a) 硅试剂的种类和特点:anti 构型(syn/anti:5:95)的产物,ee:99%产物的构型与烯醇双键的构型无关。
+RCHO PhOR O MeOH Zr cat.有机铜和锡催化剂(Evans )中心金属(铜或锡)和配体对产物立体化S-t-Bu OTMS MeOMe -78o C (S MeO O HO (2R)-anti-syn /anti (R Sn(OTf)2-11a (10 mol%)Sn(OTf)2-11b(10 mol%)-78o CMeOMeOO Me Cu(OTf)2-11c (10 mol%)NNO O MeMeRR 11b: R=Bn, 11c: R=t -Bu(S -78o C(2S)-anti-syn /anti (2S)-syn-syn /anti 发现了配体的加速作用,配体的加入极大地减小了水解反应PhPhO OH PhPh O OH +chiral cat.H 2O/C 2H 5OH(1:9)(a)手性有机金属催化剂催化直接aldol 反应的历程Trost试剂Up to 99%ee催化剂,一个Lewis酸的作一个Zn起碱的作用。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
催化导论
——不对称双羟基化反应学号:10110310班级:1011031姓名:戴明明
摘要
不对称双羟基化反应又名为夏普莱斯不对称双羟基化反应,是巴里·夏普莱斯在Upjohn双羟基化反应的基础上,于 1987 年发现的以金鸡纳碱衍生物催化的烯烃不对称双羟基化反应。
与sharpiess环氧化反应一样,该反应也是现代有机合成中最重要的反应之一。
原理
不对称二羟基化反应(asymmetric dihydroxylation,AD)是一类重要的催化不对称反应[1],它不仅是许多手性药物,如紫杉醇C13侧链、美托洛尔、普萘洛尔、氨苄青霉素、昆虫激素和维生素D活性代谢物合成中的关键步骤[2],而且还为不对称催化反应中新型配体的合成提供了必需的手性砌块[3].研究该反应的核心问题之一是手性配体的设计与合成.迄今为止,文献已经报道了许多行之有效的配体,但是有些配体制备复杂、分离难度大、价格昂贵,因此设计合成简单,价廉和高效的手性配体仍然是目前的研究重点[4].本研究以天然金鸡纳生物碱奎宁和辛可宁为原料,将其结构中的活性基团羟基转换为碱性更强的氨基,与对氯苯甲酰氯反应得到新的手性配体1和2,考察这两种配体在
AD反应中的催化活性及不对称诱导作用.
典型的反应条件是四氧化锇(OsO4)和二氢奎宁(DHQ)或二氢奎尼丁(DHQD)的手性配体衍生物作为催化剂,以计量的铁氰化钾、N-甲基吗啉N-氧化物(NMO)或叔丁基过氧化氢作为再氧化剂,并加入其他添加剂如碳酸钾和甲磺酰胺等。
现实条件中常用非挥发性的锇酸盐K2OsO2(OH)4 代替OsO4。
[8][9] 市售的二羟化混合物试剂称为AD-mix,有 AD-mix α(含(DHQ)2-PHAL)和AD-mix β(含 (DHQD)2-PHAL)两种。
大多数烯烃在上述条件下,能都以高产率、高ee值生成光学活性的邻二醇,而且反应条件温和,无需低温、无水、无氧等条件。
DHQ 和DHQD 衍生物可分别用于一对对映异构邻二醇的合成,反应产物的立体构型可根据烯烃的结构,利用下图来进行预测。
DHQ 和 DHQD 的各类衍生物都可用作催化剂的手性配体,但最有效的配体是酞嗪(2,3-二氮杂萘)衍生物(DHQ)2-PHAL 和(DHQD)2-PHAL。
反应机理
反应的催化循环如下。
由锇酸甲原位生成的四氧化锇与配体结合为络合物,该络合物对烯烃发生对映选择性的环加成反应(锇氧化反应)。
生成的锇酸脂,经水解得到二醇和被还原的锇酸盐。
后者被计量的再氧化剂氧化为四氧化锇而重复使用。
实例
奎宁;辛可宁;不对称双羟化反应
材料
奎宁,辛可宁);叠氮化钠;甲磺酰氯,对氯苯甲酰氯;甲磺酰胺,(E)二苯乙烯,苯乙烯,α甲基苯乙烯,(E)β甲基苯乙烯,β萘乙烯,锇酸钾(Aldrich 试剂);其余试剂为国产AR.或PC.级试剂,XRC1显微熔点仪),温度计未经校正;PERKINELMER 343型自动旋光仪; );INOVA400型核磁共振波谱仪),未作特殊说明者均以CDCl3为溶剂,TMS为基准物质; Apex II和ZABHS型质谱仪);III 型元素分析仪);600E型高效液相色谱仪);Chiralcel OJ, OD, OBH, AD, ODH 手性色谱柱。
方法
氨基脱氧奎宁的制备将奎宁甲磺酸酯溶于27 mL DMF中,加入NaN3 (420 mg,6.4 mmol),85~90℃搅拌21 h.向反应液中加入20 mL水,用30 mL乙酸乙酯萃取3次.有机层再用水洗,无水硫酸镁干燥.减压蒸除溶剂得红色胶状物.将上述粗产物溶解在绝对甲醇中(0.1 mmol/mL),加入催化量的10% Pd/C,室温下用高纯H2常压催化氢化直到反应完全(TLC检测).反应液经短的中性Al2O3填充柱过滤,滤去不溶物,并旋转蒸除溶剂.余物经快速柱层析(EtOAc/MeOH=10∶1)分离得淡黄色油状物9氨基脱氧奎宁,产率为82%。
手性配体1的合成将9氨基脱氧奎宁(485 mg,1.5 mmol)溶于25 mL二氯甲烷和10 mL三乙胺中,0℃下慢慢滴加对氯苯甲酰氯溶液(25 mg,1.8 mmol溶于5 mL THF中).滴加完毕,继续室温搅拌,用薄层层析跟踪反应(展开剂为乙酸乙酯∶甲醇=9∶1)直到反应完全(约需要46 h).反应完毕后,向反应液中加入25 mL二氯甲烷稀释,有机相用10%的碳酸钠水溶液洗3次(20 mL×3),无水碳酸钠干燥.蒸除溶剂后,油状粗产物经快速柱层析(EtOAc/MeOH=10∶1)得白色固体,即为手性配体1,450 mg,产率81%。
手性配体2的合成以辛可宁为原料,按照上述方法合成手性配体2.
手性配体1和2催化的烯烃不对称双羟化反应于50 mL三颈瓶中加入0.98 g (3 mmol)K3[Fe(CN)6],0.41 g(3 mmol)K2CO3,0.01 mmol配体,0.8 mg (2.0 μmol)K2OsO2(OH)4,95 mg CH3SO2NH2(1 mmol)(端烯不加),1 mmol烯烃,6 mL 水和6 mL叔丁醇,0℃搅拌反应,薄层层析检测反应进程. 反应完全后,加入1.5 g Na2SO3,搅拌1 h,加入10 mL乙酸乙酯,搅拌分层.水层用乙酸乙酯萃取(15 mL×3) ,合并有机层,用2 mol/L KOH 10mL洗涤(端烯不用KOH),再用水洗至中性,无水MgSO4干燥,减压浓缩,硅胶柱层析分离。
不对称催化反应的结果手性配体1和2在锇酸钾的存在下,对5种烯烃进行了不对称二羟化反应.结果显示,这两种新型手性配体都可以有效地催化不对称双羟化反应,对5种烯烃均表现出高的催化活性(化学产率为87%~95%)和不对称诱导作用(对映体过量值为76%~93%)。
结论
烯烃的不对称双羟化反应经过近十年的发展已取得重大发展,具有良好催化效果的配体主要是金鸡纳生物碱衍生物类手性配体.在讨论配体的构效关系时, Kolb等曾指出C9位的氧原子对于配体与四氧化锇的结合是必需的,当其被碳原子取代时,配合物稳定性会显著下降. 本研究将金鸡纳生物碱9位氧原子用氮原子取代,并在9N位引入芳香取代基,所得配合物性质稳定,在不对称双羟化反应中亦表现出良好的催化活性和对映选择性.另外,通过对配体1和2的催化反应进行比较,我们也发现喹啉环上的甲氧基有利于提高反应速度和立体选择性,这一结论与Kolb等所揭示的规律相符.
总之,本研究以价廉易得的天然金鸡纳生物碱奎宁和辛可宁为原料,以温和的反应条件简便地合成了两种适合不对称双羟化反应的手性配体,为烯烃AD 反应的配体家族增加了两个新成员,与Jacobsen等报道的类似配体对氯苯甲酸奎宁酯相比,所得手性配体1和2催化的双羟化反应化学产率高,立体选择性也提高了近10个百分点.
参考文献
[1]李月明,范青华,陈新滋.不对称有机反应[M]. 北京:化学工业出版社,2005: 93-108.
[2]郑虎.药物化学[M]. 5版,北京:人民卫生出版社,2004: 105-110. [3]Cheng SK, Zhang SY, Wang PA, et al. Homogeneous catalytic asymmetric dihydroxylation of olefins induced by an efficient and recoverable polymerbound ligand QNAQNOPEGOMe[J]. Appl Organometal Chem, 2005, 19: 975-979.
[4]张生勇,郭建全.不对称催化反应[M]. 北京:科学出版社,2002: 93-95. [5] Rper S,Franz MH,Hoffmann MR. Preparetion of enantiopure 1azabicyclo [3.2.2.]nonanes functionalized at carbon C3, from cinchonine and cinchonidine[J]. J Org Chem, 2003, 68: 4944-4946.
[6]Kolb HC,Andersson PG,Sharpless KB. Toward an understanding enantioselectivity in the osmium catalyzed asymmetric dihydroxylation Kinetics[J]. J Am Chem Soc, 1994, 116(4):1278-1291.
[7] Kolb HC, van Nieuwenhze MS, Sharpless KB.Catalytic asymmetric dihydroxylation[J]. Chem Rev, 1994, 94: 2483-2547.
[8] Jacobsen EN, Marko I, Mungall WS, et al. Asymmetric dihydroxylation via ligandaccelerated catalysis [J]. J Am Chem Soc,1988, 110(6): 1968-1970.。