药典分析方法验证和确认usp37 1225 1226
USP〈1226〉药典规程的确认

USP〈1226〉药典规程的确认<1226> VERIFICATION OF COMPENDIAL PROCEDURES药典规程的确认The intent of this chapter is to provide general information on the verification of compendial procedures that are being performed for the first time to yield acceptable results utilizing the personnel, equipment, and reagents available. This chapter is not intended for retroactive application to already successfully established laboratory procedures. The chapter Validation of Compendial Procedures<1225> provides general information on characteristics that should be considered for various test categories and on the documentation that should accompany analytical procedures submitted for inclusion in USP-NF. Verification consists of assessing selected analytical performance characteristics, such as those that are described in chapter <1225>, to generate appropriate, relevant data rather than repeating the validation process.本章节的目的是提供关于药典规程确认的信息,这些规程是第一次执行,使用可用的人员、设备、试剂,来生成可接受的结果。
药典分析方法确认与验证

通过分析含有高于和低于必需的检测水平的、已知待分析物浓度的样品,以显示检测限度足够低。例如,如果必需检测浓度在0.1%的杂质,则应当证明该分析规程将可靠地检测在这个水平的杂质。
药典分析方法确认与验证
D
精密度、线性等内容的验证,证明修订后的分析方法的合理可行。
当变更达到一定程度时,则需要完整的验证。如分析方法完全改变,则应按新方法进行完整的验证。
方法验证的一般原则:
通常情况下,分析方法需进行方法验证。对于仅需按照实验室日常测试操作步骤即可测定的检验项目不需要进行验证,如外观、崩解时限、密度、重量、pH值、灰分、装量等。
对于展示出背景噪音的仪器分析规程,ICH文件描述了一个通用方法,用来比较从以下样品测得的信号,这些样品分别为含已知低浓度被分析物的样品和空白样品。这样就确立了能够可靠检测的待分析物的最低浓度。可接受的典型信噪比是2:1或3:1。其他方法取决于校正曲线斜率的测定和响应值的标准差。
5QUANTITATION LIMIT定量限度
第一类:对成药中原料药或活性成分(包括防腐剂)的主要组分进行定量测定的分析规程。
第二类:对成药中原料药或降解物质中杂质进行测定的分析规程。这些规程包括定量测定和限度测定。
第三类:对工作特性(例如,溶出度、药物释放)进行测定的分析规程。
第四类:鉴别测试。
每一个类别均需要不同的分析信息。每个种类通常所需要的数据要素在表2中列出。
分析工作特性
第二类
第一类
定量测试
限度测试
第三类
第四类
准确度பைடு நூலகம்
USP40 1226 药典的确认中英文对照

1226 VERIFICATION OF COMPENDIAL PROCEDURES药典方法的确认The intent of this general information chapter is to provide general information on the verification of compendial procedures that are being performed for the first time to yield acceptable results utilizing the personnel, equipment, and reagents available.此章节的意图是对药典方法的确认提供基本资料,使用人员,设备和试剂使第一次进行运用药典方法以产生可接受的结果。
This chapter is not intended for retroactive application to already successfully established laboratory procedures. The chapter Validation of Compendial Procedures <1225>provides general information on characteristics that should be considered for various test categories and on the documentation that should accompany analytical procedures submitted for inclusion in USP–NF.Verification consists of assessing selected analytical performance characteristics, such as those that are described in chapter <1225>to generate appropriate, relevant data rather than repeating the validation process.此章节并不旨在对已经成功建立的实验室方法进行回顾性运用。
药典方法确认规程
目的建立药典方法确认规范,确保药典方法适用于本实验室的实验条件。
定义药典方法的确认:收载于CP、USP、BP、EP等药典的产品分析方法,使用前证明该验证过的产品测试方法、条件确实适用于本实验室的实际使用环境。
职责质量控制室所有人员负责按照本规程执行药典分析方法的确认。
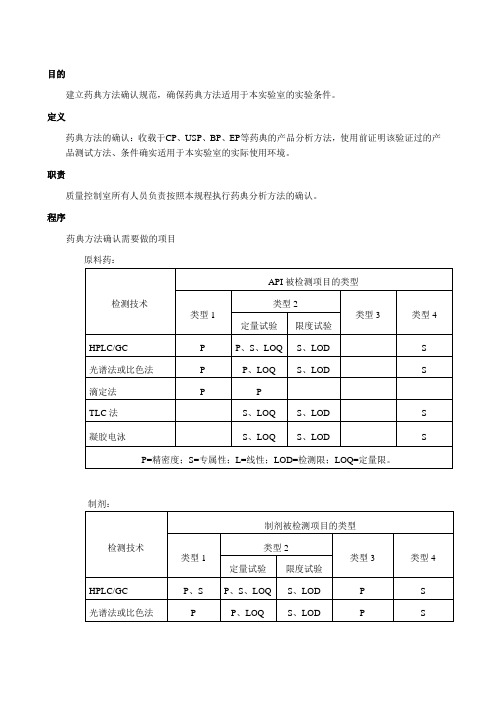
程序药典方法确认需要做的项目原料药:制剂:被检测项目类型的说明:系统适用性实验按药典正文要求的内容进行;正文没有要求的内容,如RSD%,按药典附录的要求进行。
专属性系指在其他成分(如杂质、降解物、辅料等)可能存在下,采用的分析方法能够正确鉴定、检出被分析物质的特性。
通常,鉴别、杂质检查、含量测定方法中均应考察其专属性。
如采用的方法不够专属,应采用多个方法予以补充。
鉴别反应专属性鉴别试验应确证被分析物符合其特征。
专属性试验要求证明能与可能共存的物质或结构相似化合物区分,需确证含被分析物的供试品呈正反应,而不含被测成分的阴性对照呈负反应,结构相似或组分中的有关化合物也应呈负反应。
杂质检测专属性在杂质可获得的情况下,可向供试品中加入一定量的杂质,证明杂质与共存物质能得到分离和检出,并具适当的准确度与精密度。
在杂质或降解产物不能获得的情况下,专属性可通过与另一种已证明合理但分离或检测原理不同、或具较强分辨能力的方法进行结果比较来确定。
或将供试品用强光照射,高温,高湿,酸、碱水解及氧化的方法进行破坏(制剂应考虑辅料的影响),比较破坏前后检出的杂质个数和量。
必要时可采用二极管阵列检测和质谱检测,进行色谱峰纯度检查。
含量检测专属性➢对于API主成分含量测定可在供试品中加入杂质,考察测定结果是否受干扰,并与未加杂质的供试品比较测定结果;在杂质或降解产物不能获得的情况下,可采用另一个经验证了的或药典方法进行比较,对比两种方法测定的结果。
也可采用破坏性试验(强光照射,高温,高湿,酸、碱水解及氧化),得到含有杂质或降解产物的试样,用两种方法进行含量测定,比较测定结果。
药品分析方法的验证和确认标准操作规程
目的:方法验证的目的是证明采用的方法适合相应检测要求;方法确认目的是为确认方法在本实验室条件下的适用性。
应用范围:适用于本公司产品的检验方法。
编订依据:《中国药典》2010年版二部附录194页。
内容:1 分析方法的验证及确认的适用范围及发起时机2 方法验证方法验证就是根据检验项目的要求,预先设置一定的验证内容和验证标准要求,并通过设计合理的实验来验证所采用的分析方法是否答合检验项目的要求。
建立质量标准时,应对分析方法中的各检验项目进行完整的验证;在药品生产工艺变更、制剂的组分变更、原分析方法进行修订时,可根据变更的内容决定对分析方法进行部分验证还是完整的验证;当原料药合成工艺发生变更时,可能引入新的杂质,杂质检查方法和含量测定方法的专属性就需要进行验证,以证明有关物质检查方法能够检测新引入的杂质,且新引入的杂质对主成分的含量测定应无干扰;当质量标准中某一项目分析方法发生部分改变时,如采用高效液相色谱法测定含量时,检测波长发生改变,则需要重新进行检测限、定量限、专属性、准确度、精密度、线内容的验证,证明修订后的分析方法的合理可靠;当变更达到一定程度时,则需要完整的验证。
如分析方法完全改奕,则应按新方法进行完整的验证。
2.1 方法验证的一般原则通常情况下,分析方法需进行方法验证。
对于仅需按照实验室日常测试操作步骤即可测定的检验项目不需要进行验证,如外观、崩解时限、密度、重量、pH值、灰分、装量等。
方法验证的内容应根据检验项目的要求,结合所采用分析方法的特点确定。
同一分析方法用于不同的检验项目会有不同的验证要求。
2.2 需要验证的检验项目检验项目是为控制药品的质量,保证药品安全有效而设定的测试项目。
根据检验项目的设定目的和验证内容的不同要求,一般需验证的检验项目分为四类:鉴别、杂质的限度检查、杂质的定量测定、含量测定包括原料药或制中有效成分的含量,制中其他成分的含量,溶出度与释放度等检查中的溶出量,以及含量均匀度。
药典分析方法验证和确认usp37--1225-1226

药典分析方法验证和确认usp37 1225 1226 <1225>药典分析方法验证用于评价药物质量的检验方法应符合一定要求。
根据联邦食品、药品、化妆品第501条的规定,在美国药典(USP)和国家处方集(NF)的正文检验方法和规定为法定标准。
按照现行药品生产管理规范有关条文[21 CFR 211.194(a)]要求,用于评价药品是否符合既定标准规定的分析方法应具有一定准确性和可靠性。
按照该要求[21 CFR 211.194(a)(2)],USP和NF分析方法使用者不必验证方法的准确性和可靠性,但要确认在实际使用条件下的适用性。
考虑到USP和NF标准法定地位这一基本情况,所以,药典品种新方法或修订方法得建议,应由充分实验室数据支持,以说明方法有效。
本指导通则内容尽可能与三方国际协调会(ICH)关于在欧盟、日本和美国注册申报资料要求中与分析方法部分有关的分析方法验证和方法学文本一致。
药典申报新的或修订的分析方法得药典申报,应报送充分的资料,以便美国药典委员会专家理事会及其专家委员会建议的方法是否清晰完整,需要建立该方法的结论,以及方法验证文本。
方法类型不同,申报要求可能不同,一般包括下列内容:理由写明需要本方法的理由、用途以及与其他方法比较的优点。
修订的方法应写明现行药典方法的局限性以及建议方法得优点。
建议的分析方法应完整详细地写明建议的分析方法,以便他人依法操作。
文字应包括全部重要的操作参数和有关事项以及明确的试验结果计算公式。
数据元素本部分要求提供详细、完整的分析方法验证文件,文件应包括实验数据及各适用的分析方法运行特性计算结果。
运行特性在下文中叙述。
验证分析方法验证是通过实验室研究证明该分析方法的运行特性符合其分析应用要求的过程。
表1中列出了本文分析方法验证需考虑的典型分析运行特性。
由于对术语有不同的理解和用法,本通则以下部分包括各运行特性的定义以及衡量特性的方法描述。
定义与“测定结果”有关。
分析方法验证与确认管理规程

分析方法验证与确认管理规程0 目录1 目的 (1)2 适用范围 (1)3 定义 (1)4 职责 (2)5 规程内容 (3)6 环境、健康和安全EHS............................................................................ 错误!未定义书签。
7 相关文件................................................................................................... 错误!未定义书签。
8 附件........................................................................................................... 错误!未定义书签。
9 变更历史记录........................................................................................... 错误!未定义书签。
1 目的为保证检测方法的适用性、重现性和可行性,特制订本规程。
本规程明确了检验方法验证的基本内容和测试方法,为检验方法验证、确认工作明确了基本原则。
2 适用范围2.1 对检验方法进行方法学验证时;2.2 对在研产品的检验方法进行复核确认时;2.3 对首次使用的药典、国标等检验方法进行确认时。
3 定义3.1 检验方法验证:证明采用的方法适用于相应检测要求。
3.2 检验方法确认:证明使用法定方法在目前实验室条件下是否能获得可靠结果,是否适用于相应的检测工作。
在本质上和验证一样,但不一定是验证项目的全部。
3.3 药典方法:经过国家药监部门批准的药典收载的质量标准和检验方法3.4 法定方法:法定方法包括药典方法、国标方法等。
USP 1225 - 药典规程的验证

1225药典规程的验证用于评价药品质量水平的检验规程受到多方面要求影响。
根据联邦食品、药品和化妆品法案501章,由美国药典和国家处方集各论中的含量测定和质量标准构成了法定标准。
现行GMP[21 CFR 211.194(a)]要求,用于评价药物与已建立标准符合性的检测方法,必须符合在准确度和可靠性方面的适当标准的要求。
根据这些法规[21 CFR 211.194(a)(2)],在美国药典-国家处方集中有描述的分析方法无需验证这些方法的准确度和可靠性,而仅需要核查在实际使用条件下的实用性。
认识到美国药典和国家处方集的法律地位,因此,建议采用新的或是修订的药典分析规程,以充分的实验室数据支持记录方法的有效性。
本信息章节的内容最大程度的与三方国际会议(ICH)的文件《分析规程验证》和《方法学》的延伸内容保持一致,涉及到的分析规程包括欧盟、日本和美国提交的注册申请的一部分。
药典意见书关于新的或修订的分析规程的药典意见书应该包括充足的信息,使美国药典专家理事会及其专家委员会的成员能评估拟议规程的相对优势。
大多数情况下,验证涉及到评估分析规程描述的清晰性和完整性,需要此规程的确定性,和已恰当验证过的文件。
信息可根据涉及到的方法类型而改变。
然而,大部分情况下,提交建议包括以下部分:基本原理—这部分内容应该确定规程的需要性,描述提出的此规程的性能和它优于其他类型的原因。
对于修订的规程,应提供相比之下现行药典规程的局限和拟定规程的优势。
拟定分析规程—这一部分应该充分对分析规程进行完整性描述,具体到能使业内人士重复它。
文章应包括所有重要的操作参数和具体指令,如:试剂制备、进行系统稳定性试验、所用空白的描述、预防措施、实验结果计算的明确公式。
数据要素—这一部分应提供完全的、完整的此分析规程验证的文件材料。
其应该包括对于证明每个实用分析性能特征的实验数据和计算的总结。
这些特性在下面的部分描述。
验证一个分析规程的验证是建立和实验室研究的过程,该程序的运行特性满足预定分析程序的要求。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
药典分析方法验证和确认usp37 1225 1226 <1225>药典分析方法验证用于评价药物质量的检验方法应符合一定要求。
根据联邦食品、药品、化妆品第501条的规定,在美国药典(USP)和国家处方集(NF)的正文检验方法和规定为法定标准。
按照现行药品生产管理规范有关条文[21 CFR 211.194(a)]要求,用于评价药品是否符合既定标准规定的分析方法应具有一定准确性和可靠性。
按照该要求[21 CFR 211.194(a)(2)],USP和NF分析方法使用者不必验证方法的准确性和可靠性,但要确认在实际使用条件下的适用性。
考虑到USP和NF标准法定地位这一基本情况,所以,药典品种新方法或修订方法得建议,应由充分实验室数据支持,以说明方法有效。
本指导通则内容尽可能与三方国际协调会(ICH)关于在欧盟、日本和美国注册申报资料要求中与分析方法部分有关的分析方法验证和方法学文本一致。
药典申报新的或修订的分析方法得药典申报,应报送充分的资料,以便美国药典委员会专家理事会及其专家委员会建议的方法是否清晰完整,需要建立该方法的结论,以及方法验证文本。
方法类型不同,申报要求可能不同,一般包括下列内容: 理由写明需要本方法的理由、用途以及与其他方法比较的优点。
修订的方法应写明现行药典方法的局限性以及建议方法得优点。
建议的分析方法应完整详细地写明建议的分析方法,以便他人依法操作。
文字应包括全部重要的操作参数和有关事项以及明确的试验结果计算公式。
数据元素本部分要求提供详细、完整的分析方法验证文件,文件应包括实验数据及各适用的分析方法运行特性计算结果。
运行特性在下文中叙述。
验证分析方法验证是通过实验室研究证明该分析方法的运行特性符合其分析应用要求的过程。
表1中列出了本文分析方法验证需考虑的典型分析运行特性。
由于对术语有不同的理解和用法,本通则以下部分包括各运行特性的定义以及衡量特性的方法描述。
定义与“测定结果”有关。
分析方法的叙述应写清该方法得测定结果是什么。
ISO5725-1和3534-1指出,“测定结果是用特定测定方法得到的特性值。
分析方法应规定进行一次还是数次单独测定,作为测定结果报告的平均值,或其他适宜函数(如,中位数或标准偏差)。
可能要求做标准校正,如气体体积校正到标准温度和压力时的体积。
所以测定结果可以是数个观察值的计算结果。
简单试验时,测定结果就是观察值本身”。
测定结果是用于与标准规定限度比较的最终报告值,但偶有例外。
物理性质测定方法验证可能包括化学计量模型。
方法验证中用到的典型分析特性可用于基于化学计量模型的方法。
表1 分析方法验证用典型分析特性准确度精密度专属性检测限定量限线性范围耐用性用代表性样品进行分析方法验证时,应考虑工艺条件和物料差异的影响。
下列情况下,药典分析方法应进行再验证:报送USP的修订分析方法;或将已有通用方法用于新产品或新物料制成的产品(见下文方法所需数据元素部分)。
国际协调会(ICH)有关文本建议下列情况下需在验证:原料药合成工艺改变;药物制剂组方改变;分析方法改变。
通则<1225>提供适于大多数药典分析方法得验证要求信息。
药典分析方法验证可按照表1中列出的部分或全部分析特性,以及表2中分析方法分类进行。
有的药典分析方法验证要求超出通则<1225>的特性。
则可按各有关方法通则规定的分析验证特性或特殊验证要求进行。
分析运行特性准确度定义分析方法得准确度系指该方法所得测试结果与真值的接近程度。
分析方法的准确度应在一定范围内测定。
[关于术语:通则<1225>和ICH Q2中准确度定义为不偏离程度(真实度)。
但在国际计量学词汇[International Vocabulary of Metrology(VIM)]和国际标准组织[the International Organization for Standardization(ISO)]的文件中,“准确度”有不同的意思。
ISO准确度的定义把不偏离程度(真实度)与精密度的概念合在一起]。
测定对于原料药含量测定,用分析方法测定已知纯度的供试品(如对照品),或用被验证方法测定结果与其他已知准确度的方法测定结果进行比较,测得准确度。
对于制剂中药物的含量测定,用分析方法测定制剂成分混合物中按照方法范围加入的已知量被测物,测得准确度。
如不能获得全部制剂成分,可向制剂中加入已知量被测物(加样法)进行测定,或用被验证方法测定结果与其他已知准确度的方法测定结果进行比较。
对于杂质定量分析,原料药或制剂中加入已知量杂质,测得准确度。
如不可能得到某些杂质或降解产物,可以将测定结果与另一方法测得结果进行比对。
如没有其他方法,可比较杂质和被测物质的响应值,即等量杂质和被测物响应值比值(相对响应因子),如果已知该比值,可用于计算杂质含量。
准确度为样品中被测物已知加入量含量测定的百分回收率,或平均值与可接受真值的偏差,同时报告置信区间。
ICH有关文件建议在规定范围至少3个浓度至少测定9次的结果确定准确度(即3个浓度,每个浓度重复测定3次)。
准确度的确定可以用不同方法,包括在含量测定范围内测定被测物回收率(百分回收率),或测定估计浓度与真实浓度的线性关系。
建议统计判断标准为斜率的置信区间应在约1.0区间内,或斜率应接近1.0。
在验证文件中均应规定区间或接近程度的定义。
判断标准取决于含量测定方法和变异以及结果。
不建议在斜率为1.0时用零假设检验没有统计意义地确定判断标准。
物理性质测定方法的准确度可通过标准物质分析确定,或根据具体情况对上述方法得适用性进行具体分析来确定。
精密度定义分析方法的精密度系指均匀样品多次取样用该分析方法测定各结果的一致程度。
分析方法的精密度常用一系列测定结果的标准偏差或相对标准偏差(变异系数)表示。
精密度衡量在正常操作条件下分析方法的重现性和重复性。
重复性系指不同实验室用该分析方法测定,如协同研究。
中间精密度(也称耐用性)为实验室内差异,即在同一实验室在不同日期,或用不同分析人员,或用不同设备用该分析方法进行测定。
重复性系指在一个实验室由一个分析人员用相同设备在短时间内用该分析方法进行测定。
测定分析方法的精密度是对足够份数的一个均匀样品进行测定,以便计算标准偏差或相对标准偏差(变异系数)统计有效的估计数。
本测定为各份样品从样品制备得到最终结果进行的全部分析过程。
ICH有关文件建议在规定范围至少3个浓度至少测定9次的结果确定准确度(即3个浓度,每个浓度重复测定3次),或用100%测试浓度至少6次测定结果。
专属性定义 ICH定义专属性在杂质、降解产物以及辅料存在下分析方法能准确确定被测物质存在的能力。
如果缺乏专属性,可用其他分析方法弥补。
[注:国际专业权威机构(IUPSC,AOAC-I)采用“选择性”这个术语,对选择性极高的分析方法保留“专属性”这个术语]。
对于下面讨论的分析方法,以上定义具有以下含义: 鉴别试验:鉴定被分析物特性纯度检查:采用的所有分析方法能给出被测物质中杂质的准确含量(如有关物质检查、重金属限度检查、有机挥发性杂质检查)。
含量测定:给出正确结果,样品中被分析物含量或效价的准确结果。
测定定性分析(鉴别试验)方法。
应能显示区分可能存在的结构十分相关的化合物的能力。
用含有被分析物质的阳性反应(与已知对照品对照)加上不含被测物质样品的阴性反应进行确证,并确证阳性反应不是与被分析物结构相似或十分相关的物质产生的。
杂质分析方法,向原料药或制剂中加入适量杂质,应能显示这些杂质的测定结果具有一定准确度和精密度。
含量测定分析方法,其专属性应能显示该分析方法不受杂质或辅料存在的影响。
实际做法可向原料药或制剂中加入适量杂质或辅料,确定测定结果不受这些外来物质的影响。
如果杂质或降解产物得不到,可将含有杂质或降解产物的样品测定结果与用另一确证了的方法测定结果比较(如药典方法,或其他经验证了的方法),以说明其专属性。
应包括对贮存在相应苛刻条件下的样品测定比较(如:光、热、湿度、酸或碱水解、以及氧化)。
含量测定分析方法,应比较这些结果;色潜纯度检査应比较杂质的检出情况.ICH有关文件说明,当使用色谱分析方法时,验证资料中应附代表性色谱图,以显示其选择性好坏,应标明每个色谱个峰。
峰纯度检査(如用二极管阵列或质谱法检测)可用于显示被测定物色谱峰仅为一个成分。
用波谱法的定性定量测定方法验证,应参考有关通法,如近红外光谱法、拉曼光谱法、X射线粉末衍射法等。
检测限定义检测限是限度检查的特性。
检测限系指在规定试验条件下样品中被分析物能被检出的最低量,不一定要求定量。
因此,限度检查只要说明被分析物的量髙于或低于一定限度。
检测限通常以样品中被分析物的浓度表示(如百分浓度,十亿分之几)。
测定非仪器分析方法,一般用已知浓度被分析物的样品进行分析,求得该被分析物能被可靠检出的最低浓度。
仪器分析方法,可用与非仪器分析方法相同方法求得检测限。
分析方法推荐作为药典方法时,不必要测定实际的检测限。
实际上,用含高于和低于规定检测浓度的已知浓度被分析物样品求得的检测限是非常低的。
例如,要求检査浓度为0.1%的杂质,应表明,用本法能可靠地检出该浓度的杂质。
具有基线噪音的仪器分析方法,ICH有关文件叙述了一个常用方法,即将含已知低浓度被分析物样品测得的信号与空白样品信号比较。
可以确定可靠检出被分析物的最低浓度。
一般可接受的信噪比为2:1或3:1。
其他方法如按照标准曲线的斜率和响应值的标准偏差计算检测限的。
无论用那种方法,必须用接近检测限或制备成检测限浓度的数个样品,对检测限进行最后验证。
定量限定义定量限是样品基质中低浓度化合物定量测定的特性,如原料药中的杂质和最终药物制剂中的降解产物。
定量限系指在规定试验条件下样品中被分析物能被测定的最低量,且具有可接受的精密度和准确度。
定量限通常以样品中被分析物的浓度表示(如百分浓度,十亿分之几)。
测定非仪器分析方法,一般用已知浓度被分析物的样品进行分析,求得该被分析物能被测定且具有可接受准确度和精密度的最低浓度。
仪器分析方法,可用与非仪器分析方法相同方法求得定量限。
分析方法推荐作为药典方法时,不必要测定实际的定量限。
实际上,用含高于和低于规定定量浓度的已知浓度被分析物样品求得的定量限是非常低的。
例如,要求测定被分析物的浓度为每片0.lmg,应表明,用本法能可靠地定量测定该浓度的被分析物。
具有基线噪音的仪器分析方法,ICH有关文件叙述了一个常用方法,即将含已知低浓度被分析物样品测得的信号与空白样品信号比较。
可以确定可靠测定被分析物的最低浓度。
一般可接受的信噪比为10:1。
其他方法如按照标准曲线的斜率和响应值的标准偏差计算定量限的。
无论用那种方法,必须用接近定量限或制备成定量限浓度的数个样品,对定量限进行最后验证。