主要表现为发育迟缓的儿童微缺失综合征10例
九种常见染色体微缺失

九种常见染色体微缺失1. 5p 微缺失(5p deletion syndrome):这是一种由于5号染色体短臂上的一段基因缺失引起的病症。
常见症状包括智力发育迟缓、面部异常、心脏问题和肌肉松弛等。
2. 7q 微缺失(7q deletion syndrome):这是一种由于7号染色体长臂上的一段基因缺失引起的病症。
常见症状包括智力发育迟缓、面部特征异常、心脏问题和生长发育延迟等。
3. 11q 微缺失(11q deletion syndrome):这是一种由于11号染色体长臂上的一段基因缺失引起的病症。
常见症状包括智力发育迟缓、面部异常、言语问题和心脏缺陷等。
4. 13q 微缺失(13q deletion syndrome):这是一种由于13号染色体长臂上的一段基因缺失引起的病症。
常见症状包括智力发育迟缓、面部异常、眼睛和心脏问题等。
5. 15q 微缺失(15q deletion syndrome):这是一种由于15号染色体长臂上的一段基因缺失引起的病症。
常见症状包括智力发育迟缓、面部异常、行为问题和癫痫等。
6. 16p 微缺失(16p deletion syndrome):这是一种由于16号染色体短臂上的一段基因缺失引起的病症。
常见症状包括智力发育迟缓、面部异常、言语和运动协调问题等。
7. 18p 微缺失(18p deletion syndrome):这是一种由于18号染色体短臂上的一段基因缺失引起的病症。
常见症状包括智力发育迟缓、面部异常、胎儿生长受限和心脏问题等。
8. 22q 微缺失(22q deletion syndrome):这是一种由于22号染色体长臂上的一段基因缺失引起的病症。
常见症状包括智力发育迟缓、面部异常、心脏问题、免疫系统问题和精神疾病等。
9. Xq 微缺失(Xq deletion syndrome):这是一种由于X染色体长臂上的一段基因缺失引起的病症。
常见症状包括智力发育迟缓、面部异常、性别特征异常和心脏缺陷等。
十种出生缺陷1

十种出生缺陷1十种出生缺陷1.无脑儿(1)临床症状:无脑儿是神经管畸形的一种。
无脑畸形,为大脑完全缺如,头皮、颅盖骨缺失,双眼突出,颈短,脑部发育极原始,脑髓暴露,不可能存活,仅有基底核等由纤维结缔组织覆盖,出生后婴儿无法生存,不久即死亡。
(2)发病原因:"无脑儿"是神经管畸形的一种.神经管畸形的产生,是由于在胚胎时期,受到遗传和环境中诸多因素的影响,导致神经管闭合不全,从而形成无脑畸形、脊柱裂等。
(造成胚胎神经管闭合不全的原因;1.绒毛膜促性腺激素不足,2.维生素B12或叶酸缺乏可影响胚胎神经管闭合3.妊娠剧吐或孕妇有糖尿病造成酮血症酸中毒4.孕妇高热38.5℃以上,持续超过1周5.用药不当,如孕早期服用雌激素类避孕药)(3)预防及治疗:孕期3至8周是致畸的敏感期,做好此期的保健工作是预防先天性畸形的关键。
此期的保健要点是:避免营养失调,不偏食;减少接触传染病的机会(如少去公共场所,避免接触发烧患儿等);妊娠剧吐要及时治疗;服药要遵医嘱;工作环境要尽量避免或减少接触有毒有害物品;生活起居要规律,减少精神刺激,不吸烟和酗酒2.唐氏综合征(又称21三体综合征、先天愚型)(1).发病原因:唐氏综合症是染色体结构畸变所致的疾病,卵子在减数分裂时21号染色体不分离,形成异常卵子,导致患者的核型为47,XX(XY),+21。
母亲年龄过大、过小都可能导致唐氏综合症的发生。
(2)临床特征:患者主要表现为生长发育迟缓,多发畸形及智力低下。
患者还有特殊的面容:头颅小而圆,鼻梁扁平,眼裂小,外眼角上斜,眼距宽,内眦赘皮明显;舌大常伸出口外,流涎;患者的软骨发育差,四肢较短,身材矮小,指短;肌张力低下,常有腹直肌分离或脐疝;患者手指的箕形纹多,50%的患者为通贯手,约50%的患者有先天性心脏病;男患者常有隐睾而没有生育能力,女患者通常无月经,但少数能生育。
(3)预防及治疗:避免离电辐射、避免大量用药、避免接触化学物质、避免病毒感染。
7例儿童生长发育迟缓病例分享

确诊试验
②精氨酸激发生长激素分泌试验:静脉注射精氨酸(0.25 g/mL),0.5 g/kg体重,最大剂量不超过30g。用NS溶液 3ml/kg稀释于30 min内滴完,需空腹试验。滴注前及注 完后30、60、90、120 min各取血1次测生长激素.
生长发育迟缓
生长发育迟缓是指在生长发育过程中出现速度放 慢或是顺序异常等现象。发病率在6%~8%之间。
在正常的内外环境下儿童能够正常发育,一切不 利于儿童生长发育的因素均可不同程度地影响其 发育,从而造成儿童的生长发育迟缓。
原因
1.正常的生长变异 占80%~90%,如家族性矮身材、体质性发育延
生长发育迟缓病因调查(二)
病理性原因
特发性矮小: 生长激素缺乏性矮小: 性早熟: 骨发育障碍: 宫内发育迟缓: Turner Syndrome: 其他染色体异常: 其他:
7.9% 7.9%
3.2% 2.3% 1.4% 1.4% 0.8%
5.0%
体格发育落后 运动发育落后 语言发育落后 智力发育落后 心理发育落后
智力
按对智力影响分类: 1伴智力障碍:先天型甲低,唐氏综合症,粘多糖病 2不伴智力障碍:家族性,体质性,GHD,软骨发育不
全,肾小管酸中毒等
生长发育迟缓的诊断步骤(1)
1:详细询问病史:包括患儿出生时的胎龄、分娩方式、出生 情况、母亲的妊娠及生产史,个人生长发育史和患病史、 生后及养育环境(物质及精神)等.收集患儿以往测量的身 高记录,绘制身高生长曲线。
PHIP基因突变所致Chung-Jansen综合征一例
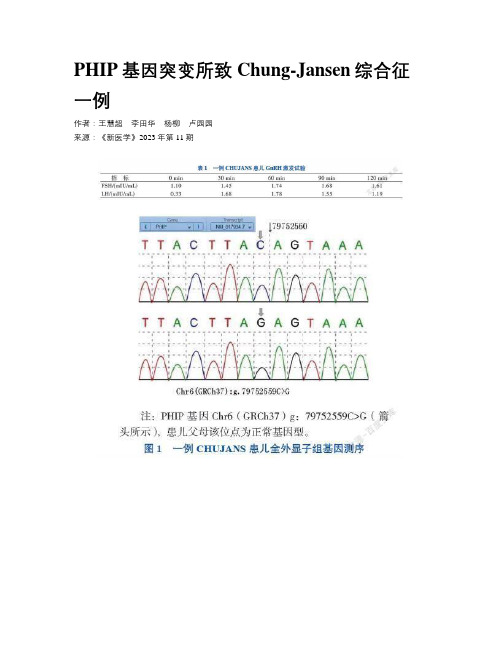
PHIP基因突变所致Chung-Jansen综合征一例作者:王慧超李田华杨柳卢园园来源:《新医学》2023年第11期【摘要】 Chung-Jansen综合征(CHUJANS)是一种常染色体显性遗传病,是新近发现的罕见肥胖综合征,主要表现为发育迟缓、智力障碍、肥胖和畸形。
该文报道1例以肥胖、睾丸小为主要表现的CHUJANS患儿,该患儿发育迟缓、智力障碍,伴有左肾缺如及低促性腺激素性性功能减退,基因检测结果提示PHIP基因突变,突变位点c.600+1G>C,最终诊断为CHUJANS。
经过长期综合性治疗,患儿远期生活质量获得极大改善。
CHUJANS发病率低,且累及多系统,该例扩展了CHUJANS的基因突变谱,有助于提高临床医师对该疾病的认识水平,及早识别并给予干预将有助于改善患者预后。
【关键词】 Chung-Jansen综合征;PHIP基因;杂合突变;儿童A case of Chung-Jansen syndrome caused by PHIP gene mutation Wang Huichao△, Li Tianhua, Yang Liu, Lu Yuanyuan. △970 Hospital of the PLA Joint Logistic Support Force,Weihai 264299, ChinaCorresponding author, Lu Yuanyuan, E-mail:*******************【Abstract】 Chung-Jansen syndrome (CHUJANS), an autosomal dominant genetic disorder, is a newly discovered rare obesity syndrome, mainly manifesting as developmental delay, mental retardation, obesity and dysmorphism. We reported one CHUJANS child with obesity and small testes as the main manifestations. The patient had developmental delay, mental retardation, complicated with left renal agenesis and hypogonadotropic hypogonadism. Genetic testing prompted PHIP gene mutation at c.600+1G>C. The child was diagnosed with CHUJANS. After long-term comprehensive treatment, the long-term quality of life was significantly improved. As Chung-Jansen syndrome is low in prevalence and multi-systemic, this case report expands the spectrum of mutations in CHUJANS,which can deepen clinicians’ understanding of this disease. Early diagnosis and intervention contribute to enhancing clinical prognosis.【Key words】 Chung-Jansen syndrome; PHIP gene; Heterozygous mutation; ChildrenChung-Jansen综合征(CHUJANS,OMIM#617991)是一种以发育迟缓、智力障碍、肥胖和畸形为特征的常染色体显性遗传病,由位于6q14染色体上的PHIP基因(OMIM#612870)中的杂合突变所致,可在婴儿期发病,大多为基因突变从头合成,少数为家族遗传[1]。
22q11.2缺失综合征一例

22q11.2缺失综合征一例摘要22q11.2缺失综合征是人类常见的染色体缺失综合征,不同患者之间的临床症状表现出高度的异质性,极易造成误诊、漏诊。
本文报道1例经全外显子组测序确诊的22q11.2缺失综合征患儿的临床特点。
女,3岁,以“不会说话,行走姿势异常”为代主诉就诊河南省儿童医院康复科。
患儿偶会发“奶奶”音,主动言语少,叫名有反应,可完成简单指令,会指认五官及日常用品,不会示意大小便。
弓背坐,不能从仰卧位直接坐起,坐位、站立、行走时头偏向左侧,行走姿势异常。
不会上下楼梯,不会独跑,不会双脚蹦离地面。
咀嚼能力欠佳,易流涎。
查体无特殊面容,头颅外观无畸形,头围47.8cm。
神经系统检查双下肢肌张力略高,病理征无异常。
辅助检查:头颅核磁示双侧额颞叶巨脑回,局部多小脑回畸形;双侧侧脑室后角旁斑点状异常,考虑脱髓鞘或髓鞘化不良;双侧侧脑室轻度扩张,双侧侧脑室周围血管间隙稍扩张。
婴幼儿感觉发育检测:视觉、听觉反应未见明显异常,前庭觉、本体觉失常。
OT:1.双上肢力量差,肌耐力不足,肩关节稳定性差,关节囊松弛;2.双侧拇示指对捏小物体不灵活,指尖捏无发完成,手指分离动作欠佳,3.双手协调性差,手指控制能力欠佳,手肌协调欠佳。
精神科B类量表:运动相当于21个月,社会适应相当于15个月,智力相当于21个月。
核型分析:46,XX。
心脏彩超、甲状腺功能、血尿遗传代谢筛查、肝肾功能、乳酸、血氨、同型半胱氨酸等均无异常。
既往体质较差,易患呼吸道感染,在外院间断康复治疗1年余,运动逐渐进步,但进步缓慢。
全外显子组测序结果显示22号染色体q11.21区域(chr22:18893887-21823635)存在2.93Mb的杂合缺失。
该区域包含蛋白编码基因55个,包括明确单倍剂量不足基因TBX1,22q11.2缺失综合征诊断明确。
入院以来,给予经颅磁刺激治疗、平衡功能训练、作业疗法、小儿捏脊治疗、低频、言语训练等综合康复治疗,患儿运动机能进步,但仍存在语言、运动和智力全面发育落后。
7号染色体缺失的案例

7号染色体缺失的案例
一种常见的7号染色体缺失的案例是威廉姆斯综合征(Williams syndrome)。
威廉姆斯综合征是一种罕见的遗传性疾病,患者通常在出生时就会出现面部特征和身体发育上的异常。
这一综合征的发病率约为1/7500至1/20000。
威廉姆斯综合征的主要特征包括智力发育迟缓、心血管问题、特殊的面部特征(如宽阔的额头、扁平的鼻梁、嘴唇较厚)以及多样化的认知和行为问题。
患者通常具有出色的音乐才能和社交能力,但在其他领域可能存在较大的学习困难。
威廉姆斯综合征是由于7号染色体的一部分缺失所导致的,具体而言是7q11.23区域的缺失。
这一缺失可能是由于染色体的重组错误或染色体突变引起的,而这一缺失又导致了患者身体和认知上的异常。
目前尚无特效的治疗方法可以彻底治愈威廉姆斯综合征,但早期的干预和治疗可以帮助患者提高生活质量和适应能力。
患者通常需要终生的医疗和教育支持,以便最大限度地发挥他们的潜力。
《人体发育学》(名词解释+简答题)

《人体发育学》一、名词解释1.生长发育:生长发育人的生长发育是指从受精卵到成人的成熟过程。
2.生长:生长是指儿童身体器官系统和身体形态上的变化,以身高(身长)、体重、头围、胸围等体格测量表示,是量的增加。
3.发育:发育是指细胞组织和器官的分化与功能成熟,主要指一系列生理、心理和社会功能发育,重点涉及儿童的感知发育、思维发育、语言发育、人格发育和学习能力的发育等,是质的改变。
4.成熟:成熟是指生命体的结构和功能成为稳定的完全发育状态,心理学的成熟是指内在自我调节机制的完成和完善状态。
5.气质:气质是人在行为方式上表现出的强度、速度、稳定性、灵活性和指向性等动态的心理特征。
6.性格:性格是指一个人对客观现实的某些态度以及与这些态度相适应的比较牢固的行为方式,是个性的核心。
7.脑性瘫痪:脑性瘫痪是自受孕开始至婴儿期脑发育阶段非进行性脑损伤和发育缺陷所导致的综合征,主要表现为运动障碍及姿势异常。
8.学习障碍:学习障碍属于特殊障碍,是指在获得和运用听、说、读、写、计算、推理等特殊技能上有明显困难,并表现有相应的多种障碍综合征。
9.胎儿期:胎儿期是人体发育的最早阶段,是婴儿出生前在母体宫内发育的阶段,从受孕到分娩共10个月左右,约280天。
10.胎动:胎动是指胎儿在母体内自发的身体活动或蠕动。
11.早产儿:早产儿胎龄不足37周的活产婴儿。
12..粗大运动发育:粗大运动发育指抬头、坐、翻身、爬、站、走、跳等运动发育,是人类最基本的姿势和移动能力的发育。
13.平衡反应:当身体重心移动或支持面倾斜时,机体为了适应重心的变化,通过调节肌张力以及躯干与四肢的代偿性动作,保持正常姿势。
14.平衡:平衡是指在不同的环境和情况下,维持身体直立姿势的能力。
15.精细运动能力:精细运动能力指个体主要凭借手以及手指等部位的小肌肉或小肌群的运动,在感知觉、注意等心理活动的配合下完成特定任务的能力。
16.手眼协调:手眼协调指在视觉配合下手的精细动作的协调性。
22q11微缺失和微重复综合征

腭裂(cleft palate,C)
低钙血症(hypocalcemia,H)
14
正常人与患者颅底比较
VCFS患者
The Faces of VCFS
长脸; 小下颌; 大鼻;球状鼻尖; 睑裂狭窄; 淡漠,缺乏面部表情
15
图片来源:
外耳畸形
16
• 两个CAFS患者的面容
眼睑浮肿、 鼻梁低平、 智力发育迟缓
Cleft palate-craniofacial journal,septemper 1997 Vol.34 No.5:425-429
32
22q11微缺失综合征:3相关综合征 CAFS
椎干异常面容综合征
• 患病率:罕见病( rare disease )
• 遗传方式:80-95%为新发病例,15-20%为家族遗传,遗
传方式为常染色体显性
• 22q11微缺失见于98-100%的患者
An orphan or rare disease is generally considered to have a prevalence of fewer than 200,000 affected individuals in the United States. (2005年统计数据显示美国人口约2.964亿,据此计算其患病率低于 1:1480 即6.8×10-4 ) /
为常染色体显性
• 疾病资料:人类孟德尔遗传在线数据库(OMIM)将其编
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
主要表现为发育迟缓的儿童微缺失综合征10例
作者:钟丽霞王鷁超
来源:《医学信息》2014年第24期
摘要:目的探讨以发育迟缓为主要表现的儿童微缺失综合征的临床特点及临床诊断思路。
方法 2009年1月~2014年1月湖南省妇幼保健院小儿康复科共有染色体微缺失10例。
对其发育评估结果、伴随症状、辅助检查等临床资料进行回顾性分析。
结果 10例患儿全都有运动、语言及智力等发育落后,6例有特殊面容及先天畸形,有的还伴有癫痫等。
确诊年龄6个月~12岁,平均39.7月。
结论儿童微缺失综合征中以发育迟缓为主要表现,且多伴先天畸形、特殊面容等。
对于发育迟缓严重且康复疗效不佳的患儿,在排除了遗传代谢病等疾病之后,还应做染色体病的相关检查。
关键词:微缺失综合征;发育迟缓;特殊面容;先天畸形;儿童
微缺失综合征是由于染色体上一些小带缺失所引起的疾病总称。
它属于一种染色体畸变,常表现为先天性多发畸形、智力、体格发育迟缓,以及流产或死胎等。
小儿康复临床工作中常见到发育迟缓严重且康复疗效不佳的患儿,临床诊断很重要。
本院小儿康复科2009年1月~2014年1月共有表现为发育迟缓的染色体微缺失10例。
现报告如下。
1资料与方法
1.1一般资料表现为发育迟缓的微缺失患儿10例。
均经微阵列比较基因组杂交技术检测确定基因组异常,男9例,女1例。
确诊年龄6个月~12岁,平均39.7个月。
1.2临床表现 10例患儿全都有运动、语言及智力发育落后,6例有特殊面容及先天畸形,特殊面容包括前额突出、眼距宽、眼裂小、鼻梁低平、伸舌、高腭弓、招风耳,先天畸形包括左右小腿发育不对称、隐睾、漏斗胸、先天性心脏病(房间隔缺损)等,1例伴有高热惊厥,1例伴有癫痫,2例伴有听力差,2例伴有视力差,4例伴有体格生长发育落后,3例伴有孤独症谱系障碍。
1.3辅助检查 8例染色体核型分析为46,XY,1例为46,XX,1例为47,XY,+1mar。
基因芯片结果分别为:chrXp2
2.33缺失,chrYp22.11缺失,chr7q11.22缺失,chr10q21.1缺失,chr15q11.2-q1
3.2缺失,chr11p15.4-15.5重复,ch22q13.3-qter缺失,chrXq25缺失,
chr4P16.3缺失,chr11P15.5-P15.4重复,chr2q15-14缺失。
2讨论
儿童发育迟缓(developmental delay,DD)是发育性残疾的一种,特指
在小儿康复科临床工作中常见的一过性的单项发育落后在早期发现早期干预后一般疗效较好,对于有些症状严重的疗效不佳的发育迟缓,常见病因有遗传代谢病、染色体病、大脑发育不良和神经肌肉疾病等。
患遗传性代谢病的婴幼儿以运动、智力发育落后、肌张力低下等较常见。
韩连书等[5]随访30例检出有基因突变的甲基丙二酸血症患儿,存活的22例患儿中14例遗留不同程度运动、语言发育迟缓及智力低下。
程静[6]等发现84例Citrin蛋白缺陷所致新生儿肝内胆汁淤积症患儿中有48例合并精神运动发育迟缓。
Horster等[7]报道的273例单纯型甲基丙二酸血症患者中54%遗留不同程度运动发育迟缓。
大脑发育不良也是发育迟缓的常见病因之一,分为原发性和继发性。
杜鹃[8]等在74例原发性单纯性胼胝体发育不良的患儿中发现生长发育迟缓的占47.31%,智力低下占33.78%。
刘毅生[9]等研究结果表明:脑损伤所致的脑白质髓鞘化异常、缺氧缺血性脑病后遗改变、皮层发育异常、胼胝体发育不良、脑畸形是婴幼儿运动发育迟缓的主要颅内原因。
神经肌肉疾病指运动单位疾病,排除由于脑部病变引起的肌肉功能异常。
儿童时期运动单位病常见。
婴幼儿神经肌肉疾病的常见临床表现为全身肌张力低下和运动发育迟缓[10]。
以上10个病例均排除了遗传代谢病和神经肌肉病,以上病例中有一例为脑白质发育不良,也可能为继发于染色体病所致。
所有病例经微阵列比较基因组杂交技术检测结果均异常。
这给我们的启示如下:对于临床中有些症状严重且疗效不佳的发育迟缓患儿,在排除了遗传代谢病、神经肌肉疾病、大脑发育不良之后,还应做染色体病的相关检查。
染色体病是由于染色体畸变即数目异常和(或)结构畸变所引起的疾病。
无论是数目异常还是结构畸变,其实质是涉及染色体或染色体节段上基因群的增减或位置的转移,使遗传物质发生了改变,都可以导致染色体异常综合征或染色体病。
据统计,人类中新生儿的染色体异常发生率为0.5%~1%。
微缺失综合征是由于染色体上一些小带缺失所引起的疾病,和21-三体综合征、18-三体综合征等都属临床常见的常染色体综合征。
传统的细胞遗传学分析技术能检测出象21-三体综合征、18-三体综合征等染色体病,染色体显带技术结果虽然准确可靠,但分辨率低,难以检测小于5 Mb的CNVs,更难以确定CNVs 的大小和断裂点,且需要细胞培养、显微计数和分析,费时费力。
较小范围的染色体不平衡畸变引起的智力低下、器官畸形和生长发育落后的患者群体中,常规G显带技术检出的异常率只有15%~ 40%,漏诊率高达60%~85%。
更细小的染色体片段异常可通过高分辨染色体分析或FISH检测确定。
Hochstenbach 等总结了29 篇应用微阵列技术来检测MR/DD 的文献,发现微阵列技术作为首选方法检出率可达19%,是传统细胞遗传学方法的2 倍。
Cooper 等分析15767 例智力低下儿童的基因组DNA,发现14.2%的患儿由>400 Kb 的基因组不平衡导致患儿
发病。
微阵列比较基因组杂交(array.CGH)具有高分辨率、高灵敏度、高通量、自动化和快速等优点,能准确地检测微缺失、微复制和扩增等基因组不平衡,尽管目前尚存在费用和技术方面的问题,但array.CGH可以作为G显带染色体分析的有益补充应用于临床细胞遗传诊断中。
综上,总结如下:①对于临床中发育迟缓的患儿在排除了遗传代谢病等病因后要多从染色体病角度查找病因。
②能在亚显微水平对整个基因组进行分析,是G显带染色体分析的有益补充。
③要做好染色体病所致发育迟缓的优生优育咨询工作,进行早期预防和早期诊断,降低患病儿的发生,提高人口素质。
在此要特别感谢广州金域医学检验中心细胞遗传学实验室的孟亚仙老师为此文提供的指导。
参考文献:
[1]The New York City.Department of Health.Developmental Delay in Young Children:Identification and Intervention [Z].CM E Activity Inside,2001.
[2]Shevell M,Ashwal S,Donley D,et a1.Practice parameter:Evaluation of the child with global developmental delay[J].Neurology,2003,2:367-380.
[3]Stein MT.Unraveling the causes of global developmental delay[J].Arch Dis Child,2007,92:l81-182.
[4]Srour M,Mazer B,Shevell MI.Analysis of clinical features predicting etiologic yield in the assessment of global developmental delay[J].Pediatrics,2006,118(1):118-139.
[5]韩连书,毋盛,楠叶军.单纯型甲基丙二酸血症患者诊治分析[J].中华医学遗传学杂志,2013,30(5):589-593.
[6]程静,刘丽,盛慧英,Citrin蛋白缺陷所致新生儿肝内胆汁淤积症84例分析[J].广东医学,2012,33(17):2559-2562
[7]Horster F.Garbade SF.Zwickler T,et a1. Prediction of outcome in is01ated methylmalonic acidurias:combined use of clinical and biochemical parameters[J].J Inherit Metab Dis,2009,32:630-639.
[8]杜鹃,沈璐,廖伟华.胼胝体发育不良临床、磁共振成像分析(附1 1 6例)[J].卒中与神经疾病,2010,17(1):28-31.
[9]刘毅生,沈家亮,杨思达.婴幼儿运动发育迟缓的头颅MRI表现及病因分析[J].实用医学影像杂志,2010,11(5):273-276.
[10]Richard E.Behrman,Robert M.Kliegman,Hal B.Jenson.尼尔森儿科学[M].北京大学医学出版社,2007:2605-2606.编辑/肖慧。