第七章_化学反应动力学基础1
高中化学竞赛第7章_化学动力学基础课件

0 BB B
已知 d dnB B
转化速率的定义为:
•
d
1 dnB
dt B dt
2022/4/16
反应速率(rate of reaction)
IUPAC推荐反应速率用单位体积内的转化速率表示,
反应的速率定义为:
r
1
d
V dt
( d 1 dnB ) dt B dt
1 dnB /V 1 d c B
动力学认为:
1 2
N2
3 2
H2
NH3(g)
需一定的T,p和催化剂
1 H 2 2 O 2 H 2O(l)
点火,加温或催化剂
2022/4/16
7.1 化学动力学的任务和目的
化学动力学发展简史
•1848年 van’t Hoff
dlnKc U
提出:
dT RT2
dlnkEa dT RT2
Kc
kf kb
例如:
r k 0
r k 反 应
r k [ A ] [ B ] 二 级 , 对 A 和 B 各 为 一 级
r k [ A ] 2 [ B ] 三 级 , 对 A 为 二 级 , 对 B 为 一 级
r k [ A ] [ B ] - 2
负 一 级 反 应
r k [ A ] [ B ] 1 /2
k 的单位随着反应级数的不同而不同。
2022/4/16
准级数反应(pseudo order reaction)
在速率方程中,若某一物质的浓度远远大于其 他反应物的浓度,或是出现在速率方程中的催化 剂浓度项,在反应过程中可以认为没有变化,可 并入速率系数项,这时反应总级数可相应下降, 下降后的级数称为准级数反应。例如:
化工-第七章 化学反应动力学基础

反应速率与转化率:
设A组分:n A0 初始量、t反应时间、n At时刻瞬时量 x At时刻瞬时转化率 反应消耗的A的量 n A0 n A xA 反应初始时A的量 n A0 即: n A n A0 (1 x A ) 若反应前后体积变化不大:c A c A0 (1 xA ) 1 dnA 1 nA0 dx A 则: rA V dt V dt nA0 x A
转化为目的产物的反应物的物质的量 选择性()= 反应物被转化掉的物质的量 收率:
收率()= 转化为目的产物的反应物的物质的量 进入反应器的反应物的物质的量
二、复杂反应的速率方程式
1、平行反应:
k2 A B S dcS dcP a1 b1 a b 则:rp k1c A cB rS k2c A2 cB2 dt dt rp k1 a1 a2 b1 b2 平行反应速率之比为: = c A cB rS k2 k1 A B P
第七章 化学反应动力学基础
内容: 2、简单反应的速率方程式 4、本征动力学和宏观动力学
1、化学动力学基本概念 3、简单反应和复杂反应
重点: 2、简单反应和复杂反应
1、简单反应的速率方程式
§7-1 化学动力学基本概念
一、化学计量方程式
复杂的化学计量方程式: 0= i Bi
n
i : 为组分Bi的计量系数。反应物为负、产物为正。
r f (c, T ) r f (T ) (c) f (T ):反应速率的温度效应、 (c):反应速率的浓度效应 f (T )常表示为反应速率常数k : k A exp( E 对于均相反应:aA bB sS
( c) c cB A
化学动力学

x tcA0(cA0
x)
1 cA
1
cA0
1 cA0
x
1
cA0
kAt 或kA
cA0 cA tcA0cA
x tcA0(cA0
x)
反应的特征:
1 cA 对 t 作图是一条直线,斜率即 kA 。
kA 具有浓度1·时间1的量纲,表达为L3N1T1。
半衰期与 kA 和 cA0 的乘积成反比。
t1/ 2
• 反应级数 (n) • 速率系数 (k)
(mol m3 )1n s1
k的物理意义是当物质的浓度均为单位浓度时 k 等于 反应速率,因此它的数值与反应物的浓度无关。在催 化剂等其它条件确定时,k 的数值仅是温度的函数。
k 的单位随着反应级数的不同而不同。
复合反应:
幂函数型速率方程
=
k
c
A
c
B
适用于玻色子组成的独立子系统。 同种粒子间相互不可区别,多个粒子可以具有相同量子态。
费米-狄拉克分布(FD分布)
适用于费米子组成的独立子系统。 同种粒子间相互不可区别,每个量子态最多只有一个粒子。
麦克斯韦–玻尔兹曼分布
Nj
g e j /(kT ) j
N
q
q
g e i /(kT )
ii
子配分函数
微观状态的量子力学描述
(1)平动能级:粒子在容器中的平
移运动所具有的能量
平动能级:公式题中会给出
z xy
t
h2 8m
nx2
l
2 x
n2y
l
2 y
nz2 lz2
;
nx , ny , nz为正整数
微观状态的量子力学描述
07 第七章 均相反应动力学基础

二、复合反应的速率方程式
1. 平行反应
k1 A B P 主反应 k2 A B S 副反应
a1 b1 rP k1cA cB a2 b2 rS k2cA cB
rP k1 a1 a2 b1 b2 cA cB rS k2
ρ :选择率
(1)ρ增大,反应的选择性增大
2. cA,0 ≠ cB,0 cA ≠ cB
rA=kcAcB
cA,0xA = cB,0xB
rA k (cA,0 cA,0 xA )(cB,0 cB,0 xB ) kcA,0 (1 xA )(cB,0 cA,0 xA )
cB,0 令: M rA kcA,0 2 (1 xA )( M xA ) cA,0
ln ln 1 xB cA,0 ( M 1)kt 1 xA 1 xB (cB,0 cA,0 )kt 1 xA
p11,例7-1
三、一级可逆反应
垐 1垎 A噲 垐P k
2
k
A的反应速率为正、逆反应速率的代数和:rA = k1cA-k2cP
rA k1cA k2cP ( k1 k2 )cA,0 ( xA,e xA )
(1)φ(c) 用幂级数的形式表示: (c ) cAcB (2)α,β为反应物A,B的反应级数 (3) α+β= n称为反应总级数。反应级数越高,浓度的变化对反应 速率的影响越显著 (4)基元反应,α,β与计量系数相等 (5)非基元反应,α,β由实验测定,可为整数或分数
四、反应速率的温度效应f(T) (p7)
nA = nA,0(1 - xA,0)
cA = cA,0(1 - xA,0) 1 dnA 1 nA,0dxA 用转化率表示反应速率:rA V dt V dt nA,0 xA 用转化率表示反应进度: νA
第七章 复相反应动力学

e Ea ( ) / RT
f
( )
式中:亦称为凝聚系数,代表具有Ea能量的吸附质分子碰撞在空白吸附
中心上而被吸附中心吸附的分子的概率;通常,由于固体表面的不均匀性
及被吸附的吸附质分子间的相互作用,Ea、均院
第三节 吸附过程动力学
3. 脱附速率rd基本方程
ra rd ka pA (1A ) kdA
令 bA ka / kd ,称为吸附平衡常数或吸附系数;Langmuir吸附等温式:
A
bA pA 1 bA pA
19
东南大学能源与环境学院
第三节 吸附过程动力学
b. 单组份吸附,吸附时,吸附质分子发生解离,每个吸附质
占据2个以上吸附中心:
7
东南大学能源与环境学院
第一节 概述
③ 固体表面的分子与聚集到固体表面的反应物首先形成一种不 稳定的中间络合物,然后不稳定的中间络合物反应得到反应 产物,相当于化学吸附作用;
④ 反应物在固体表面形成一种所谓“表面自由基”,然后按链 式反应机理进行。
无论采用何种解释,共同点在于对表面反应的关键步骤的确定
2.吸附
不同相态物质接触时,一相分子(吸附 质)只停留在相界面上,吸附量与相际 表面积关系密切,遵循朗格缪尔、乔姆 金或弗鲁德里希等吸附模型。
因气相很易进入液相内部,故气液接触 时,还常称为吸收;气相、液相分子不 易进入固相内部(不包括进入固体内表 面空隙),故气固、液固间传质过程通 常称为吸附。
① 反应物分子借助于固体表面富集,固体表面依靠其表面分 子的分子间力(范德华力),把周围邻近的分子拉到固体 表面上浓集,相当于物理吸附作用;
② 固体表面凭借其表面所存在的作用力(分子间力、化学键 力),使靠近固体表面的反应物分子发生变形,使反应物 分子反应能力发生改变,致使反应得以进行或加速,相当 于物理吸附、化学吸附的双重作用;
第七章 配合物反应动力学
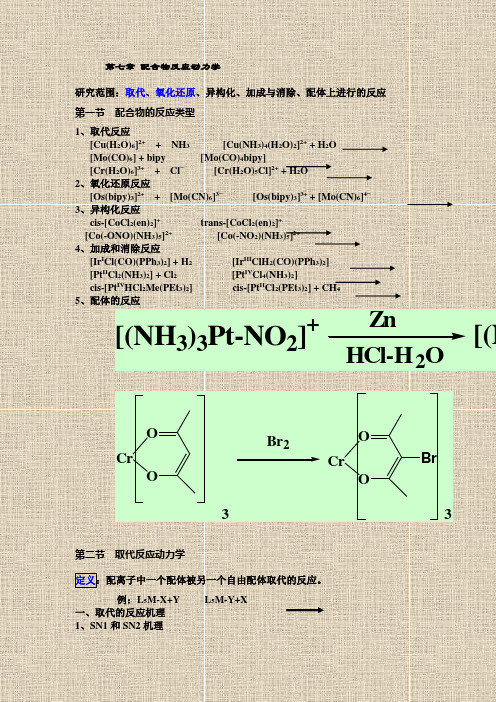
第七章配合物反应动力学研究范围:取代、氧化还原、异构化、加成与消除、配体上进行的反应第一节配合物的反应类型1、取代反应[Cu(H2O)6]2++ NH3 [Cu(NH3)4(H2O)2]2+ + H2O[Mo(CO)6] + bipy [Mo(CO)4bipy][Cr(H2O)6]3++ Cl−[Cr(H2O)5Cl]2+ + H2O2、氧化还原反应[Os(bipy)3]2++ [Mo(CN)6]3−[Os(bipy)3]3+ + [Mo(CN)6]4−3、异构化反应cis-[CoCl2(en)2]+trans-[CoCl2(en)2]+[Co(-ONO)(NH3)5]2+[Co(-NO2)(NH3)5]2+4、加成和消除反应[Ir I Cl(CO)(PPh3)2] + H2[Ir III ClH2(CO)(PPh3)2][Pt II Cl2(NH3)2] + Cl2[Pt IV Cl4(NH3)2]cis-[Pt IV HCl2Me(PEt3)2] cis-[Pt II Cl2(PEt3)2] + CH45、配体的反应ZnHCl-H2O [(NH3)3Pt-NO2]+[(NCrOOCrOOBrBr233第二节取代反应动力学定义:配离子中一个配体被另一个自由配体取代的反应。
例:L5M-X+Y L5M-Y+X一、取代的反应机理1、SN1和SN2机理(1)离解机理(SN1机理)慢a.L5M-X = L5M + X(配位数下降6 5)b.L5M + Y = L5M-Y速率方程:d[L5M-Y]/dt = k[L5M-X]速率与Y浓度无关,是对[L5M-X]的一级反应。
(2)缔合机理(SN2机理)慢a、L5M-X + Y = L5MXY(配位数升高6 7)b、L5MXY = L5M-Y + Xd[L5M-Y]/dt = k[L5M-X][Y]属于二级反应。
* SN1和SN2是两种极限情況。
二.活性与惰性配合物及理论解释1、活性与惰性配合物1)定义:配体可被快速取代的配合物,称为活性配合物;配体取代缓慢的配合物,称为惰性配合物划分标准:配合物与反应试剂(浓度均为0.1M)在25℃时反应,t1/2>1min,称为惰性配合物;t1/2<1min,称为活性配合物。
第七章 化学反应动力学
第七章化学反应动力学一.基本要求1.掌握化学动力学中的一些基本概念,如速率的定义、反应级数、速率系数、基元反应、质量作用定律和反应机理等。
2.掌握具有简单级数反应的共同特点,特别是一级反应和a = b的二级反应的特点。
学会利用实验数据判断反应的级数,能熟练地利用速率方程计算速率系数和半衰期等。
3.了解温度对反应速率的影响,掌握Arrhenius经验式的4种表达形式,学会运用Arrhenius经验式计算反应的活化能。
4.掌握典型的对峙、平行、连续和链反应等复杂反应的特点,学会用合理的近似方法(速控步法、稳态近似和平衡假设),从反应机理推导速率方程。
学会从表观速率系数获得表观活化能与基元反应活化能之间的关系。
5.了解碰撞理论和过渡态理论的基本内容,会利用两个理论来计算一些简单反应的速率系数,掌握活化能与阈能之间的关系。
了解碰撞理论和过渡态理论的优缺点。
6.了解催化反应中的一些基本概念,了解酶催化反应的特点和催化剂之所以能改变反应速率的本质。
7.了解光化学反应的基本定律、光化学平衡与热化学平衡的区别,了解光敏剂、量子产率和化学发光等光化反应的一些基本概念。
二.把握学习要点的建议化学动力学的基本原理与热力学不同,它没有以定律的形式出现,而是表现为一种经验规律,反应的速率方程要靠实验来测定。
又由于测定的实验条件限制,同一个反应用不同的方法测定,可能会得到不同的速率方程,所以使得反应速率方程有许多不同的形式,使动力学的处理变得比较复杂。
反应级数是用幂函数型的动力学方程的指数和来表示的。
由于动力学方程既有幂函数型,又有非幂函数型,所以对于幂函数型的动力学方程,反应级数可能有整数(包括正数、负数和零)、分数(包括正分数和负分数)或小数之分。
对于非幂函数型的动力学方程,就无法用简单的数字来表现其级数。
对于初学者,要求能掌握具有简单级数的反应,主要是一级反应、a = b的二级反应和零级反应的动力学处理方法及其特点。
第七章化学动力学
第七章化学动力学主要内容1.化学动力学的任务和目的2.化学反应速率的定义3.化学反应的速率方程4.具有简单级数的反应5.几种典型的复杂反应6.温度对反应速率的影响7.链反应 重点1.重点掌握化学反应速率、反应速率常数及反应级数的概念2.重点掌握一级和二级反应的速率方程及其应用3.重点掌握复杂反应的特征,了解处理对行反应、平行反应和连串反应的动力学方法。
4.重点理解阿罗尼乌斯方程的意义并会应用。
明确活化能及指前因子的定义 难点1.通过实验建立速率方程的方法2.稳态近似法、平衡近似法及控制步骤的概念及其运用3.复杂反应的特征及其有关计算 教学方式1.采用CAI 课件与黑板讲授相结合的教学方式。
2.合理运用问题教学或项目教学的教学方法。
教学过程第7.1节化学动力学研究的内容和方法热力学讨论了化学反应的方向和限度,从而解决了化学反应的可能性问题,但实践经验告诉我们,在热力学上判断极有可能发生的化学反应,实际上却不一定发生。
例如合成氨的反应,223()3()2()N g H g NH g ,在298.15K 时,按热力学的结论,在标准状态下此反应是可以自发进行的,然而人们却无法在常温常压下合成氨。
但这并不说明热力学的讨论是错误泊,实际上豆科植物就能在常温常压下合成氨,只是目前还不能按工业化的方式实现,这说明化学反应还存在一个可行性的问题。
因此,要全面了解化学反应的问题,就必须了解化学变化的反应途径----反应机理,必须引入时间变量。
研究化学反应的速率和各种影响反应速率的因素,这就是化学动力学要讨论的主要内容。
一、化学热力学的研究对象和局限性:研究化学变化的方向、能达到的最大限度以及外界条件对平衡的影响。
化学热力学只能预测反应的可能性,但无法预料反应能否发生?反应的速率如何?反应的机理如何?例如:热力学只能判断这两个反应都能发生,但如何使它发生,热力学无法回答。
二、化学动力学的研究对象 化学动力学研究化学反应的速率和反应的机理以及温度、压力、催化剂、溶剂和光照等外界因素对反应速率的影响,把热力学的反应可能性变为现实性。
5 第七章 化学反应动力学基础
A P S
k1 k2
假设每一步反应都是一级反应,则
dc rA k1 c A dt dcP rP k1 c A k 2 c P dt dcS rS k2c P dt
反应开始时
c A c A,0
cP 0
k1t
cS 0
c A c A, 0 e
E愈大,反应速率对温度就愈敏感。
k A exp E RT
E 1 ln k ln A R T
lnk
E1 1 ln k1 ln A1 R T
2
1
E2 1 ln k2 ln A2 R T
E1>E2
o
1 T
例如, E=4l.87 J/mol 0℃时,为使反应速率提高一倍,需将反应温度提高11℃。 E=167,500 J/mol 0℃,提高3℃,反应速率提高一倍。 (3)E一定,同一反应,温度越低,反应速度对温度就 越敏感 例如,E=4l.87 J/mol 0℃ 为使反应速率提高一倍需将反应温度提高11℃ 1000℃ 提高273℃
dnA
A
dnB
B
dnS
S
dnR
R
ni ni 0 ξ νi
dnA=dξ· A,
1 d A V dt rA
r 1 d V dt
(3)反应转化率
组 份A反 应 掉 的 摩 尔 数 xA 组 份A的 起 始 摩 尔 数
xA
n A, 0 n A n A, 0
有机物的二聚反应:如乙烯、丙稀、异丁烯及环戊二烯的 二聚反应等; 加成反应:烯烃的加成反应等; NaClO3的分解,乙酸乙酯的皂化,碘化氢、甲醛的热分解 等。
第七章化学动力学主要内容1.化学动力学的任务和目的2.化学反应
第七章化学动力学主要内容1. 化学动力学的任务和目的2. 化学反应速率的定义3. 化学反应的速率方程4. 具有简单级数的反应5. 几种典型的复杂反应6. 温度对反应速率的影响7. 链反应重点1. 重点掌握化学反应速率、反应速率常数及反应级数的概念2. 重点掌握一级和二级反应的速率方程及其应用3. 重点掌握复杂反应的特征,了解处理对行反应、平行反应和连串反应的动力学方法。
4. 重点理解阿罗尼乌斯方程的意义并会应用。
明确活化能及指前因子的定义难点1. 通过实验建立速率方程的方法2. 稳态近似法、平衡近似法及控制步骤的概念及其运用3. 复杂反应的特征及其有关计算教学方式1. 采用CAI课件与黑板讲授相结合的教学方式。
2. 合理运用问题教学或项目教学的教学方法。
教学过程第7.1节 化学动力学研究的内容和方法热力学讨论了化学反应的方向和限度,从而解决了化学反应的可能性问题,但实践经验告诉我们,在热力学上判断极有可能发生的化学反应,实际上却不一定发生。
例如合成氨的反应,223()3()2()N g H g NH g + ,在298.15K 时,按热力学的结论,在标准状态下此反应是可以自发进行的,然而人们却无法在常温常压下合成氨。
但这并不说明热力学的讨论是错误泊,实际上豆科植物就能在常温常压下合成氨,只是目前还不能按工业化的方式实现,这说明化学反应还存在一个可行性的问题。
因此,要全面了解化学反应的问题,就必须了解化学变化的反应途径----反应机理,必须引入时间变量。
研究化学反应的速率和各种影响反应速率的因素,这就是化学动力学要讨论的主要内容。
一、化学热力学的研究对象和局限性:研究化学变化的方向、能达到的最大限度以及外界条件对平衡的影响。
化学热力学只能预测反应的可能性,但无法预料反应能否发生?反应的速率如何?反应的机理如何?例如:热力学只能判断这两个反应都能发生,但如何使它发生,热力学无法回答。
二、化学动力学的研究对象化学动力学研究化学反应的速率和反应的机理以及温度、压力、催化剂、溶剂和光照等外界因素对反应速率的影响,把热力学的反应可能性变为现实性。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
k = A • exp(−Ea/RT ) Ea k
酶催化——生物体内普遍存在的催化反应。 当底物与酶的活性基团处于一定的相应空间 位置时,两者形成中间活化物,反应历程的 变化,降低了活化能,加快了反应速率。可 用以下锁匙模型解释。
课后作业
• 7.2,7.3,7.7,7.8 7.11,7.13,7.15
cC
ν= -
1 a
d(A)= dt
1 b
d(B) dt =
= k (A)a (B)b
1 d(C) c dt
非基元反应的速率方程 一
•
比较复杂。浓度的方次和反应物的系数般不
一定相符,须由实验测定。
要
S2−O8d2(−S+2O382I−−) dt
=
2SO42− + I3− k(S2O82−)(I−)
由 实 验
CO(g) + NO2(g) → CO2(g) + NO(g) ν= − d(NdtO2) = − d(dCt O) = k(CO)(NO2)
对反应物
对总反应
反应
一 级
− dd(At ) = k(A)1 lg (A) 对t作图呈直线
二 − dd(tB) = k(B)2 级 1/(B) 对t作图呈直线 三 − dd(tC) = k(C)3 级 1/(C)2 对t作图呈直线
•过渡状态理论 •(transition state theory)
A + BC → A···B···C → AB + C 反应物 活化络合物 产物
过渡态和始态 的位能差就是 活化能,或者 说活化络合物 的最低能量与 反应物分子最 低能量之差为 活化能。
•活化能的物理意义
The activation energy (Ea) is the minimum amount of energy required to initiate a chemical reaction.
化学平衡和反 应速率是化学 反应问题的两 大不可分割的 方面,均十分 重要。
化学热力学判断常温常压下反应能进行, 且转化率很高。
实际反应速率太慢,毫无工业价值。
化学动力学的任务
• 化学反应的速率问题 • 化学反应的机理问题
化学反应速率的含义及其表示法
• 反应速率的定义
➢单位时间内反应物或生成物浓度改变量的
[H2O2]/(mol•dm-3)
0.40
0.20 0.10
v /(mol•dm-3•min-1) 0.014 0.0075 0.0038
ν=-
d(H2O2) dt
=
k(H2O2)
反应速率常数
CO(g) + NO2(g) → CO2(g) + NO(g)
CO NO2 v0 CO NO2 v0 CO NO2 v0
v = Z⋅f⋅P = Z⋅P⋅exp(-Ec/RT) = Z0 (A) (B) ⋅P⋅exp(-Ec/RT) = k (A)(B)
lg k = lg (Z0·P) – Ec/2.30RT
k 与反应分子的质量、大小、温度、 活化能、碰撞的方位等因素有关
碰撞理论把分子当作刚性球体,而 忽略了分子的内部结构。
(S2O82−)0
(I−)
− dS2O82−/确定dt
mol·dm-3
mol·dm-3 mol·dm-3·s-1
0.038
0.060
1.4 × 10-5
0.076
0.060
2.8 × 10-5
0.076
0.030
1.4 × 10-5
反应级数及各级反应的特点
• 反应的级数 2N2O(g) −A−u→ O2(g) + 2N2(g) ➢速率方程式里浓度的方次 ➢由实验测定 ☺一级、二级、三级以及零级反应等
H2 + I2 → 2HI 实验事实: − dd(Ht 2)= k(H2)(I2)
用波长578nm光照可加速反
应速率,且该波长的光只能
反应机理:
使I2分子解离。 I2 2I (快)
K
2I + H2 → 2HI (慢) k
− dd(Ht 2) = k(H2)(I)2
= kK(H2)(I2)
H2 + Br2 → 2HBr
0.10 0.10 0.005 0.10 0.20 0.010 0.10 0.30 0.015 0.20 0.10 0.010 0.20 0.20 0.020 0.20 0.30 0.030 0.30 0.10 0.015 0.30 0.20 0.030 0.30 0.30 0.045 0.40 0.10 0.020 0.40 0.20 0.040 0.40 0.30 0.060
d(HdCt l) = k(H2)(Cl2)1/2
室温下暗处反应速率极慢,加热并加
光照反应剧烈,瞬间即可完成。
反应机理: Cl2
2Cl
(链引发)
Cl + H2 → HCl + H (链增长)
H + Cl2 → HCl + Cl (链增长)
Cl + Cl → Cl2 链式反应d(HdCt l)H
+ =
③ 解:
t1/2 = ?
① lg (C2H5Cl) = lg (C2H5Cl)0 -
kt 2.303
= lg (0.40) - 2.5010-3 8 60
2.303
= - 0.92
(C2H5Cl) = 0.12 mol·dm-3
例 已知 300K,C2H5Cl 一级反应的 k=2.50×10-3 min-1 ,(A)0=0.40 mol·dm-3
ν=-
d(NO2) dt
=-
d(CO) dt
= k(CO)(NO2)
基元反应
非基元反应
反应物通过一步反应变成生成物 反应物通过多步,才能转 变成生成物。
基元反应的质量作用定律
• 质量作用定律
➢在恒温条件下,基元反应的反应速率与反 应物浓度的乘积成正比,各浓度的方次也 与反应物的系数相一致。
aA + bB
从平均速率到瞬时速率
平均速率
v = − Δ(H2O2)
Δt
v = lim −Δ(H2O2)
Δt → 0 Δt
= − d(H2O2)
dt
瞬时速率
用反应物浓度变化和用生成物浓度变化 表示的差异
——现行国际单位制的建议
aA + bB
cC + dD
ν= -
1 a
d(A)= dt
1 b
d(B) dt =
1 c
d(C) dt =
1 d(D) d dt
按此规定,一个化学反应就只有一个反应速率值。
✓实际测定的是净反应速率 ✓初速率(0) ✓可逆反应达平衡状态时,正向反应速率与逆向反
应速率相等,净反应速率等于零。
浓度与反应速率
——微分速率方程和反应级数
H2O2(aq) →I- H2O(l) + 1/2O2(g) 室温
HI(g) 的分解反应速率
反应级数 ? 半衰期(t1/2)
NOTES
➢Rate laws are always determined experimentally.
➢Reaction order is always defined in terms of reactant (not product) concentrations.
H+H→ Cl → HCl
k(H2)(Cl) =
H2 (链终止) Kk(H2)(Cl2)1/2
催化作用
2H2O2 (aq)
2H2O (l) + O2 (g)
加入KI
催化剂的特点
• 催化剂的特点: ➢显著改变反应速率; ➢反应前后自身的组成与数量保持不变; ➢不影响反应的平衡位置。
N2 (g) + 2Fe(s) → 2N-Fe(s) 2N-Fe(s) + 3H2(g) → 2NH3(g) + 2Fe(s) N2 (g) + 3H2 (g) → 2NH3 (g)
正值。一般用v表示。
H2O2(aq) →I- H2O(l) + 1/2·dm-3
Δ(H2O2)
Δt
0
0.80
20
0.40
0.40/20=0.020
40
0.20
0.20/20=0.010
60
0.10
0.10/20=0.0050
80
0.050
0.050/20=0.0025
第七章 化学反应速率
7.1 化学反应速率的意义 7.2 浓度与反应速率 7.3 反应级数 7.4 温度与反应速率 ·活化能 7.5 反应机理 7.6 催化作用
什么是化学动力学
瞬间完成的炸药爆炸反应
大西洋底泰坦尼克号 船首的腐蚀过程
?
合成氨反应
N2(g) + 3H2(g) → 2NH3(g)
ΔrGmө(298) = −33 kJ⋅mol−1 Kpө = 6.1 × 105 (298 K )
温度与反应速率
• 活化能与反应速率理论
在不同温度下反应2N2O5 → 4NO2+O2的速率常数
Arrhenius反应速率经验公式(Arrhenius方程) lg k = A + B T
lgKØ= - ΔH x 1 + C 2.30R T
lgk = - Ea x 1 + C 2.30R T
- Ea
速率常数、反应级数、活化能、中间产物 等。 ➢综合实验结果,参考理论,利用经验规则 推测反应历程。 ➢再经过多方面推敲
2NO (g) + O2 (g) → 2NO2 (g)