恶性嗜铬细胞瘤的治疗
嗜铬细胞瘤诊治进展

嗜铬细胞瘤诊治进展背景介绍嗜铬细胞瘤是一种罕见的神经内分泌肿瘤,通常起源于肾上腺或副交感神经系统的嗜铬细胞。
它们可以分泌儿茶酚胺激素(如肾上腺素和去甲肾上腺素),导致高血压,心动过速和出汗等症状。
因为嗜铬细胞瘤的症状和其他常见疾病非常相似,如高血压和焦虑症,因此它们的诊断和治疗一直是一个挑战。
诊断进展最近的研究表明,许多嗜铬细胞瘤与遗传突变相关联,如多发性内分泌腺瘤和神经纤维瘤病。
因此,家族史是嗜铬细胞瘤疑似患者应该询问的重要问题之一。
现在,诊断嗜铬细胞瘤的关键是通过测量尿液中儿茶酚胺代谢物的含量来检测。
这种检查可以检测到大约90%的嗜铬细胞瘤,但是它有一些缺陷,例如低敏感性和不良特异性。
最近,一些新技术被开发用于诊断嗜铬细胞瘤。
例如,底物类胶体传感器(SBP)和电化学传感器(e-sensor)可以在病人血液或尿液中检测到高灵敏度、高特异性的儿茶酚胺代谢物。
治疗进展嗜铬细胞瘤的治疗目标是完全摘除肿瘤,但该疾病常常处于恶性状态并会迁移到其他部位。
手术切除有助于大多数患者,但对于转移性嗜铬细胞瘤患者,手术可能不是最佳选择。
现在,一些新的治疗方法被开发用于治疗嗜铬细胞瘤。
例如,放射性核素治疗(RNT)和放射性树脂微球治疗(SIRT)可以用于治疗嗜铬细胞瘤的转移性病变。
此外,融合蛋白靶向治疗和免疫疗法也是当前研究的热点。
嗜铬细胞瘤是一种难于诊断和治疗的神经内分泌肿瘤。
最近的研究发展了一些新技术和治疗方法,为嗜铬细胞瘤患者提供了更好的设施。
尽管如此,更多的研究仍然需要开展,以便更好地理解嗜铬细胞瘤的发病机制和治疗方法的有效性和安全性。
嗜铬细胞瘤的用药选择

嗜铬细胞瘤起源于肾上腺髓质、交感神经节或其他部位的嗜铬组织,瘤体持续或间断地释放大量儿茶酚胺,引起持续性或阵发性高血压和代谢紊乱。以20~50岁多见,约有10%为恶性肿瘤。位于肾上腺者约占80%~90%,大多为一侧,少数为双侧。肾上腺外嗜铬细胞瘤称为副神经节瘤,主要位于腹部、腹主动脉旁,还可发生在肾门、肝门区等部位,也可位于膀胱内或直肠后等部位。肾上腺髓质内的嗜铬细胞瘤和主动脉旁嗜铬体可产生去甲肾上腺素和肾上腺素,肾上腺外的嗜铬细胞瘤只产生去甲肾上腺素,导致以高血压为主要表现的临床症群。
1.2代谢紊乱:肾上腺素可作用于中枢神经及交感神经系统控制下的代谢过程,引起代谢亢进的表现如发热、出汗、消瘦、心跳加快、基础代谢率增高等,肝糖原分解加速可引起血糖升高、糖耐量减低;脂肪分解加速可致血游离脂肪酸增高;儿茶酚胺促使钾离子进入细胞内可出现低钾血症。
1.3其他临床表现:可出现肠蠕动减弱,引起便秘;膀胱内的嗜铬细胞瘤患者在排尿时常引起高血压发作;肾上腺外的肿瘤部分可在腹部触及肿块,按压时可使血压骤升;嗜铬细胞瘤还可伴发与一些因基因突变而致的遗传性疾病如2型多发性内分泌腺瘤病、1型多发性神经纤维瘤等。
2药物
2.1 α受体阻断药能使血管扩张,降低血压。酚苄明(氧苯苄胺):作用时间长,半衰期约36小时,开始每次服10mg,每日2次,根据血压情况加量至有效剂量,一般每日30~60mg。酚妥拉明1~5mg静脉缓慢注射,或5~10mg溶于250~500ml液体中静滴。不良反应有直立性低血压、心动过速等。
2.2选择性α1受体阻断药可避免α受体阻断药的不良后果,如明显的低血压和心动过速等。半衰期较短,可较灵活调节用量。哌唑嗪起始口服每次0.5mg或1mg,每日2~3次,根据病情可适当加量至每次2~3mg。多沙唑嗪每日用量约2~6mg,控释片每日4~8mg。起始用小剂量可以避免发生严重的体位性低血压。
肾上腺嗜铬细胞瘤

肾上腺嗜铬细胞瘤肾上腺嗜铬细胞瘤:探索病因、症状与治疗在临床上,肾上腺嗜铬细胞瘤是一种罕见的神经内分泌肿瘤,它来源于肾上腺髓质中的嗜铬细胞。
本文将对肾上腺嗜铬细胞瘤的病因、症状以及治疗进行探讨。
一、病因肾上腺嗜铬细胞瘤是由肾上腺髓质的嗜铬细胞发生恶性肿瘤而引起的。
有研究表明,肾上腺嗜铬细胞瘤与家族遗传有一定的关系,大约10-15%的患者具有遗传性。
其中最常见的遗传病例是孤立家族遗传性肾上腺髓质增生症(PGL)。
与之相关的基因突变主要包括SDHB、SDHD和SDHC。
此外,肾上腺嗜铬细胞瘤也可以发生在孤立的散发性病例中,病因尚不明确。
二、症状肾上腺嗜铬细胞瘤具有多样化的症状,且症状难以准确诊断。
根据患者体内嗜铬细胞分泌的激素种类和水平,症状表现差异较大。
常见的症状包括持续性高血压、头痛、心悸、多汗、体重减轻、颈部的放射性酸胀疼痛以及手指震颤等。
此外,部分患者可能还会出现心动过速、心绞痛、恶心、呕吐、短暂性的中风症状以及骨骼疼痛等。
三、诊断临床上,通过一系列的检查来诊断肾上腺嗜铬细胞瘤是至关重要的。
常规检查包括测量患者的血压、心电图、血液检查和尿液检查。
同时,测量血和尿中的嗜铬细胞分泌物(如肾上腺素、去甲肾上腺素、多巴胺等)有助于确诊和判断瘤体的活跃程度。
影像学检查也是常用的辅助手段,如CT、MRI和MIBG扫描。
对于家族病例,遗传学检查也非常重要。
四、治疗肾上腺嗜铬细胞瘤的治疗方法因患者的病情而异。
对于良性小肿瘤,手术切除是首选的治疗方法。
患者在手术前需要恰当地准备,包括血压控制、激素替代治疗和液体支持等。
手术切除的目标是完全切除瘤体,以防止恶性转化和复发。
对于无法手术切除的患者,放疗和化疗可能成为其他治疗选择。
在术后的恢复期间,患者需要接受长期的随访和监测。
这包括定期检查血压、检测肾上腺激素水平以及进行影像学检查。
如有必要,还需进行重复手术,以去除复发或转移病灶。
在术前和术后的处理中,患者还可以接受靶向治疗。
嗜铬细胞瘤的病因治疗与预防

嗜铬细胞瘤的病因治疗与预防嗜铬细胞瘤是由嗜铬细胞形成的肿瘤。
肾上腺外的嗜铬细胞瘤可发生在从颈动脉到盆腔的任何部位。
可导致血压异常(常表现为高血压)和代谢紊乱综合征。
有些患者可能会因长期高血压而导致严重的心脏、大脑和肾脏损伤或严重的突然高血压而危及生命。
然而,如果能及时、早期地诊断和治疗,这也是一种可治愈的继发性高血压疾病。
嗜铬细胞瘤在高血压患者中的发病率为0.05%~0.2%发病高峰为20~50岁。
嗜铬细胞瘤占肾上腺的80%%~90%,多为一侧性;肾上腺外肿瘤主要位于腹膜外和腹主动脉旁。
多良性,恶性占10%%..像大多数肿瘤一样,散发性嗜铬细胞瘤的病因尚不清楚。
家族性嗜铬细胞瘤与遗传有关。
本病以20~大多数40岁的中青年患者几乎等于男性和女性。
其主要症状是高血压和基础代谢的变化:高血压可以是阵发性的、持续的或持续的高血压阵发性的。
持续的人通常有头晕、头痛、胸闷、胸痛、心跳心悸、视力模糊、精神紧张、焦虑、怕热等。
阵发性突然严重头痛、心悸、胸闷、苍白、出汗、呼吸急促,患者感到濒死。
此时,如果血压测量可达40.OkPa(200~300㎜Hg),大约半小时后,它可能会自行缓解。
恢复后,就像普通人一样。
在未来,一些刺激会再次发作。
逐渐发作更频繁,间隔缩短,情况也越来越严重。
发作刺激可能不是很强烈,有漱口刷牙或梦发作醒来,出汗,有濒死感。
还有巨大的肿瘤、高血压和无发作症状,或无肿块、无发作,死于其他疾病的手术。
因此,有这种症状的患者应尽快进行检查、诊断和治疗。
嗜铬细胞瘤的一般测试和检查如下:实验室检查:1.测定血尿茶酚胺及其代谢物。
2.药理学试验:分为刺激和抑制试验。
其它辅助检查:1.肾上腺CT首选扫描CT检查时,由于姿势变化或静脉造影剂注射可诱发高血压,应首先使用α-肾上腺素能受体阻断剂控制高血压,并在扫描过程中随时准备酚妥拉明。
2.磁共振显像(MRI)肿瘤与周围组织的解剖关系和结构特征具有较高的诊断价值。
嗜铬细胞瘤怎样治疗?

嗜铬细胞瘤怎样治疗?*导读:本文向您详细介绍嗜铬细胞瘤的治疗方法,治疗嗜铬细胞瘤常用的西医疗法和中医疗法。
嗜铬细胞瘤应该吃什么药。
*嗜铬细胞瘤怎么治疗?*一、西医*1、治疗嗜铬细胞瘤一旦确诊并定位,应及时切除肿瘤,否则有肿瘤突然分泌大量CA、引起高血压危象的潜在危险。
在早期,诊断多依靠临床特点及腹膜后注气造影等不够准确的技术,手术也多以剖腹探查为主,因而诊断错误及手术失败者居多。
近年来,随着生化试验及显像技术的发展,PHEO的定性和定位诊断技术大为提高,术前处理加强摘除PHEO的手术成功率得以提高。
术前应采用α受体阻滞药使血压下降,减轻心脏负荷,并使原来缩减的血管容量扩大,以保证手术的成功。
1.药物治疗(1)PHEO的定性及定位的诊断一旦明确,应立即用药物控制,以防出现高血压急症。
主要用药为长效α受体阻滞药,包括酚苄明(phenoxybenzamine)10~20mg,2次/d;哌唑嗪(prazosin)1~2mg,2次/d。
(2)合并高血压急症时可静脉给以酚妥拉明(phentolamine)。
如疗效不好可静脉输注硝普钠。
(3)如合并窦性心动过速和(或)室上性心动过速心绞痛,可口服选择性β1受体阻滞药,如美托洛尔(bataloc)和阿替洛尔(atendol)等,但在PHEO患者应用该药时,必须与α受体阻滞药合用,否则单独应用β受体阻滞药可能由于抑制了E的血管扩张作用而使血压明显升高,如用普萘洛尔(propranolol)等非选择性β受体阻滞药则升高血压的不良反应更为明显。
(4)如合并室性心动过速静脉输注利多卡因(lidocaine)。
(5)拉贝洛尔(labetalol)为一种α和β受体阻滞药,因其以β受体阻滞药为主,故也可引起血压升高,PHEO时是否应用尚有争论。
2.术前准备和药物治疗(1)α-肾上腺素能受体阻断剂:①酚妥拉明(phentolamine,Regitine):用于高血压的鉴别诊断(Regitine试验),治疗高血压危险发作或手术中控制血压,而不适于长期治疗。
嗜铬细胞瘤

嗜铬细胞瘤治疗
手术治疗 1、术前准备 (1)a肾上腺受体阻滞剂:至少2周,小剂量 开始,逐渐递加,手术前1天止。 (2) ß肾上腺受体阻滞剂:不能单独使用, 只能在使用a-肾上腺受体阻滞剂后使用。 (3)适当补充血容量。
嗜铬细胞瘤治疗
2、手术条件 血压控制到正常或大致正常(120/80±) 阵发性高血压停止 高代谢症状改善 体重增加、出汗减少 血容量恢复(红细胞压积<45%) 酚妥拉明滴注血压不再下降
病例二 尸检记录:后腹膜主 动脉旁可见一直经为 7.5cm的肿块,重约 150g,包膜完整,切 面实性质软,可见小 灶出血。 病理诊断:腹膜后副 神经节瘤伴囊性变。
嗜铬细胞瘤
定义:起源于肾上腺髓质、交感神经节 或其他部位的嗜铬组织,持续或间断地 释放大量儿茶酚胺,引起持续性或阵发 性高血压、交感神经兴奋和代谢紊乱; 但为可治愈性继发性高血压。
性较高,对于肾上腺肿瘤的种类较为困难。
MRI:无放射线,可用于妊娠者,有助于鉴别
嗜铬细胞瘤和肾上腺皮质肿瘤。
B超:无创,方便,初筛,直径>1cm的肾上腺
肿瘤定位,鉴别肿瘤质地。
131I间碘苄胍扫描:显示分泌CA的肿瘤、转移
灶、复发灶、肾上腺外肿瘤。
嗜铬细胞瘤CT
嗜铬细胞瘤MRI
嗜铬细胞瘤诊断
恶性嗜铬细胞瘤诊断:
病例一
入院诊断:心肌炎(考虑病毒性心肌炎可能性大) 治疗:吸氧,抗感染,硝普钠减轻心脏负荷(急 诊有过高血压),护肝,病情稍有好转。 体征:血压100-110/60-70mmHg,心率100-120次/ 分,出汗多(1天换几套衣服)。 检查结果:BR:WBC:14.4*10^9/L,N80.7%巨 细胞病毒:弱阳性。肝酶及心肌酶仍高。 治疗:停用硝普钠(血压稳定后),减慢心率, 护肝,补气强心,加强抗感染。 病情发展:停用硝普钠约8小时后出现血压突然升 高(220-230/130-140mmHg)。患者血压波动范 围大,伴有心悸、乏力,且大量出汗无好转。
嗜铬细胞瘤的诊治

嗜铬细胞瘤:起源于肾上腺髓质产生儿茶酚胺的嗜铬细胞肿 瘤
副神经节瘤:肾上腺外沿交感及副交感神经节分布的嗜铬组 织肿瘤
肾上腺髓质增生:临床表现与嗜铬细胞瘤相似,确诊依靠病 理学检查
高儿茶酚胺血症
副神经节瘤 ( Paragangliomas PGL)
腹主动脉旁(约10%~15%)、肾门、肾上极、肝 门区、肝-下腔静脉之间、胰头部、髂窝或附 近、血管旁(直肠后、卵巢、膀胱内)
MRI
MIBG
诊断PCC的敏感性/特异性分别为 85-88%和 70100%
131I-MIBG具有治疗恶性PPGL的作用 转移性、复发性PPGL、位于颅底和颈部、胸腔、
膀胱PGL,与SDHx基因相关PPGL检出敏感性较低 拟交感神经药、阻断CA转运药物如可卡因和三环
类抗抑郁药、钙同等阻滞剂、α及β肾上腺素能 受体阻滞剂可减少123I-MIBG浓聚,需停药2周
似),若无阳性发现,继以颈部和胸部扫描 儿童也可采用腹部超声 在大多数病例的定位诊断过程中联合使用CT和MR已经足够 MIBG: If CT/MRI negative
Large adrenal tumor paraganglioma PET SRS
良恶性鉴别困难:病理
根据病理切片上的组织形态来判定很困难 瘤细胞的形态异常不能作为良恶性的诊断依据 镜下呈恶性改变,临床却表现为良性 有的瘤细胞呈良性形态,但在术后1~15年内复发 在良、恶性的肿瘤细胞中都可看到重的嗜铬性颗粒、
症状不典型且多变:提高警惕!
传统观念认为:超过一半的患者有高血压发作或危象 实际上:大多数嗜铬细胞瘤是临床上没有料到的(特别是非
内分泌科医师),甚至在与肿瘤相关的致命后果发生时 美国Mayo Clinic尸检:证实的PCC患者中,生前从未被怀疑
氩氦刀治疗肾上腺恶性嗜铬细胞瘤
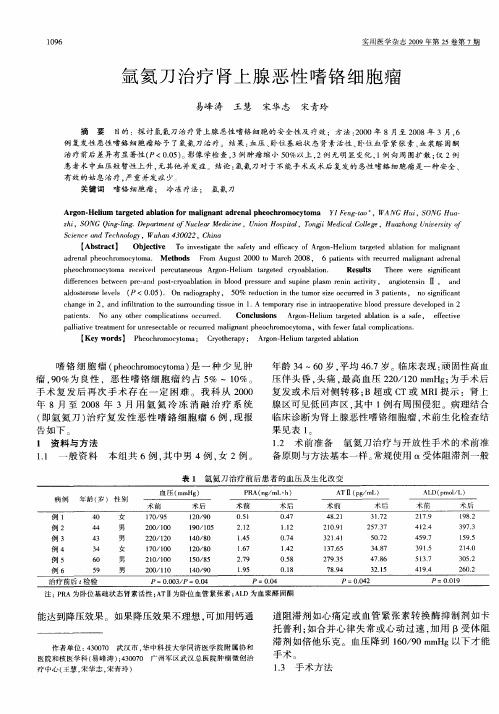
【 yw rs P ecrm ctm ; Cyteay A gnH l m t gt bai Ke od 】 h oho oyo a ro rp ; ro— eu re da l o h i a e tn
嗜 铬 细 胞 瘤 ( h oho oyo ) 一 种 少 见 肿 p e crm ct ma 是 瘤 ,0 9 %为 良性 ,恶 性 嗜 铬 细 胞 瘤 约 占 5 ~ 1 %。 % 0 手 术 复 发 后 再 次 手 术 存 在 一 定 困 难 。 我 科 从 20 00 年 8月 至 2 0 0 8年 3月 用 氩 氦 冷 冻 消 融 治 疗 系 统 ( 即氩 氦 刀 ) 疗 复 发 性 恶 性 嗜 铬 细 胞 瘤 6例 , 报 治 现
c a g n 2,a d i flr t ot e s r u di is e i A e p r r ie i n ro r ie b o d p e s r e e o d i h n e i n n tai t h uro n ngts u n 1. tm o ay rs n i ta peat l o r s u e d v lpe n 2 i on v
S inc nd T hn lg ce ea ec o o y,W u a 00 2,Chi h n 43 2 na
【 bta t A src】
0bet e T netaetesft ad e cc fA gnH l m t gt b t nfrm l nn jc v oi sgt h ae n f ayo r — ei re d al i o a ga t i v i y i o u a e ao i
p litv r amen ru e e tbl rr c re lg an he c r mo y o a l ie te t a tf nr s ca e o e u d ma in tp o b o c t ma,wi e tf tlc m p ia in . o t f we aa o lc to s h
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
Malignancy requires evidence of metastases at non-chromaffin sites distant from that of the primary tumor.
Metastatic disease in pheochromocytoma may be present at the time of initial diagnosis or may only became evident after surgical removal of the primary tumor, usually within 5 years, but sometimes 16 or more years later.
Literature Report
Therapy of Malignant Pheochromocytoma 恶性嗜铬细胞瘤的治疗
Introduction
rule of 10s for pheochromocytoma (PCC)
10% bilateral
10% extra-adrenal
10% extra-abdomen
Due to the rarity of the tumor, clinical studies about pheochromocytoma suffer from a fragmented nature and usually involve too small a number of cases to reach conclusive results.
Malignant vs. Benign
Currently, there is no effective cure for malignant pheochromocytoma.
There are also no reliable histopathological methods for distinguishing benign from malignant tumors.
Size does not reliably predict malignancy in pheochromocytomas with local disease only
Tumor size (mean ± SD) <2 cm 2.0-3.9 cm 4.0-5.9 cm 6.0-7.9 cm 8.0-9.9 cm ≥10 cm
Without treatment, the 5-year survival is generally less than 50%.
The course, however, can be highly variable with occasional patients living more than 20 years after diagnosis.
Primary surgical resection is the treatment of choice whenever possible
Limited disease: curative intention
Extended disease: still to be considered in the first place for debulking and as palliative treatment
10% malignant
10% familial来自10% children
10% normal blood pressure
Introduction
The most frequent site of metastases is the skeleton
Additional sites are liver, retroperitoneum with lymph nodes, CNS, pleura, and kidney
Should pheochromocytoma size influence surgical approach?
A comparison of 90 malignant and 60 benign pheochromocytomas
(Wen T. Shen et al.2004)
Comparison of tumor size for benign pheochromocytomas and malignant pheochromocytomas with local disease only
Once malignancy is diagnosed, therapy is generally directed at controlling blood pressure, but may also include tumor debulking.
Alternative of Current Therapy
Because there is currently no effective cure for malignant pheochromocytoma, most treatment are palliative, but in some cases may reduce tumor burden and prolong survival.
131 I-MIBG is of certain therapeutic effectiveness of symptomatic improvement
Complete tumor mass disappearance has only been found in small tumors
Treatment with 131 I-MIBG should be instituted immediately after surgical resection to eradicate the residual tumor cells and to prevent recurrences
Malignant (n = 29)
6.1 ± 3.1 cm 0 9 6 5 5 4
Benign (n = 55)
5.3 ± 2.3 cm 1 10 25 13 3 3
Malignant PCCs presenting with only local disease cannot be discriminated from benign PCCs by size alone.
Laparoscopic adrenalectomy for pheochromocytoma should be converted to open adrenalectomy for difficult dissection, invasion, adhesions, or surgeon inexperience
Surgical approach
Transabdominal approach is necessary minimally invasive procedures retroperitoneal approaches should be
abandoned to definitely preserve the tumor capsule and
(ZHU Ruisen et al. 1999)
Patients were classified into 3 groups according to their tumor size
< 8 cm3 (11 cases) , 8~20cm3 (21 cases) , > 20 cm3 (26 cases)
In group 1, the mean absorption dose per gram of tumor was above 1 000 cGy. After treatment ,tumors disappeared or shrinked in all patients
In group 2 , the absorption dose was similar to that of group 1, but the mean absorption dose per gram was 717.6 cGy , and tumor mass regression was 36 % ;76 % reduced urinary catecholamine
impossible Other treatment modalities have to be
considered
Alternative of Current Therapy
Surgery Radiopharmaceuticals Combined Chemotherapy Arterial Embolization
Surgery Radiopharmaceuticals Combined Chemotherapy Arterial Embolization
Alternative of Current Therapy
Surgery Radiopharmaceuticals Combined Chemotherapy Arterial Embolization
Bone marrow suppression is temporary and not dosage related
In 1997, Loh et al. published a review of the worldwide experience involving 116 patients treated with 131I-MIBG for malignant pheochromocytoma.