560例乳腺癌患者全基因组测序的体细胞突变图谱
全基因组测序在肿瘤领域的应用现状与未来趋势

全基因组测序在肿瘤领域的应用现状与未来趋势
佚名
【期刊名称】《中国肿瘤临床》
【年(卷),期】2024(51)1
【摘要】数十年间,传统肿瘤治疗手段均是“一刀切”,而忽视了驱动肿瘤发生发展的基因变异。
1990年发起的“人类基因组计划”,以及新一代测序技术的出现,加速了癌症基因组学的发展。
癌症基因组图谱(TCGA)计划于2005年提出,标志着癌症基因组测序工作的开始。
预计到2030年,进行基因组测序的肿瘤患者数量可达到上亿。
在肿瘤领域,临床中的基因组测序目前是利用外显子组进行测序,但外显子仅占人类基因组的1.2%。
【总页数】1页(P26-26)
【正文语种】中文
【中图分类】R73
【相关文献】
1.微生物全基因组测序在预防医学领域的应用
2.山羊内源性鼻内肿瘤病毒enENTV-FJ1株全基因组测序及生物信息学分析
3.我国首次完成牧草领域第一个全基因组测序
4.基于限制性内切酶的简化基因组测序在家禽基因组研究中的应用现状
5.全基因组重测序在畜禽研究领域的应用进展
因版权原因,仅展示原文概要,查看原文内容请购买。
乳腺癌基因突变类型

乳腺癌基因突变类型
乳腺癌基因突变类型主要包括以下几种:
1. BRCA1 和 BRCA2 基因突变:这两种基因突变是乳腺癌中最常见的基因突变类型之一,与乳腺癌和卵巢癌的发生风险增加有关。
2. P53 基因突变:P53 基因是一种抑癌基因,其突变与乳腺癌的发生和发展有关。
3. PTEN 基因突变:PTEN 基因是一种抑癌基因,其突变与乳腺癌的发生和发展有关。
4. HER2 基因突变:HER2 基因是一种癌基因,其突变与乳腺癌的发生和发展有关,特别是 HER2 阳性乳腺癌。
5. Ki-67 基因突变:Ki-67 基因是一种与细胞增殖有关的基因,其突变与乳腺癌的发生和发展有关。
以上是乳腺癌中常见的基因突变类型,不同的基因突变类型可能与乳腺癌的不同亚型和预后有关。
对于有
乳腺癌家族史的人群,可以进行基因检测,以便及早发现基因突变并采取相应的预防和治疗措施。
二代测序检测乳腺癌PIK3CA、TP53、PTEN基因突变及其临床意义

[作者简介]陆琦然(1994-02~),女,江苏东台人,硕士研究生,研究方向:乳腺癌诊疗。
E-mail:1063671621@ [通讯作者]孙洁(1985-12~),女,江苏苏州人,硕士,主治医师,研究方向:甲状腺和乳腺疾病诊治。
E-mail:sunjie_soochow@二代测序检测乳腺癌PIK3CA 、TP53、PTEN 基因突变及其临床意义陆琦然刘建夏於恩桥孙洁苏州大学附属第一医院甲状腺乳腺外科(江苏苏州215000)·临床论著·Clinical Article ·530乳腺癌是女性中发病率最高的肿瘤,也是女性死亡率最高的癌症(15.0%)[1]。
近年来精准医疗理念越发深入人心,从基因层面上进一步了解乳腺癌成为近几年医学发展的热门。
这要求科学不断发展,以追求越来越精准的基因检测,于是近年来第二代测序(next-generation sequencing,NGS)应运而生, NGS技术具备速度快、通量高及敏感度高等优势,大大弥补了第一代测序sanger法的不足[2]。
磷脂酰肌醇激酶-3催化亚基α基因(phosphoinositide-3-kinase catalytic alpha polypeptide gene,PIK3CA)是最近数十年来在乳腺癌中发现的一种高突变率原癌基因。
其突变可导致不同水平的多种信号通路的异常调控,从而通过原位杂交促进癌细胞的增殖和存活[3]。
已经有研究证明,PIK3CA基因的突变打开了乳腺癌PI3K/AKT/mTOR这条“旁门左道”,可能导致人表皮生长因子受体-2(human epidermal growth factor receptor-2,HER-2)过表达患者的靶向治疗和激素受体阳性患者的内分泌治疗出现耐药[4]。
肿瘤蛋白p53基因(TP53)基即P53基因,由11个外显子组成,是最早开始研究的几个抑癌基因之一,可诱发细胞凋亡,主导细胞分化[5]。
癌症TCGA数据库中乳腺癌预后数据的挖掘

癌症TCGA数据库中乳腺癌预后数据的挖掘Mian Khizar Hayat;王铭裕;李硕磊【摘要】近年来,乳腺癌发病率逐渐上升,并且呈现出年轻化趋势.使用TCGA数据库中已有的基因信息筛选鉴定出与乳腺癌预后相关的基因.为排除癌组织和正常组织取样时间不同造成的差异,我们选取了113对同时检测乳腺癌区和其相对应癌旁正常组织的样品,从TCGA数据库调取转录组数据,对这些数据通过DEseq进行差异表达分析,筛选出1428个差异表达基因.对差异表达基因进行基因本体GO,代谢通路KEGG,疾病本体DO和富集分析获得68个与乳腺癌相关的差异表达的关键基因;采用数据库中所用癌症的表达数据(共1097例)对这些乳腺癌相关基因进行总生存率分析,筛选出8个与乳腺癌预后相关的基因.结果显示在乳腺癌病人中PGLYRP2、SEMA3G、PROL1及SLC7A3的高表达伴随着乳腺癌病人的预后良好,而SKA1、BIRC5、RRM2和AURKA基因的高表达伴随着乳腺癌病人的预后不良.这8个基因有可能是乳腺癌预后相关的重要基因,这为乳腺癌病人的预后治疗提供了新的方向与思路,并可能通过调控基因水平来尽可能地控制预后.【期刊名称】《生物学杂志》【年(卷),期】2018(035)004【总页数】5页(P62-66)【关键词】癌症基因组图谱数据库;乳腺癌;差异表达基因;预后【作者】Mian Khizar Hayat;王铭裕;李硕磊【作者单位】兰州大学生命科学学院生物物理所,兰州730000;兰州大学生命科学学院生物物理所,兰州730000;兰州大学生命科学学院生物物理所,兰州730000【正文语种】中文【中图分类】R737.9乳腺癌是危害女性身心健康的最主要的恶性肿瘤,男性乳腺癌患者比较少见,Cancer Statistics 在 2017 年的统计数据显示乳腺癌在女性癌症发病中占据了 30%的比例[1]。
近年来,乳腺癌的发病率逐年上升,并且年轻化趋势明显[2]。
全基因组重测序数据分析详细说明

全基因组重测序数据分析1. 简介(Introduction)通过高通量测序识别发现de novo的somatic和germ line 突变,结构变异-SNV,包括重排突变(deletioin, duplication 以及copy number variation)以及SNP的座位;针对重排突变和SNP的功能性进行综合分析;我们将分析基因功能(包括miRNA),重组率(Recombination)情况,杂合性缺失(LOH)以及进化选择与mutation之间的关系;以及这些关系将怎样使得在disease(cancer)genome中的mutation 产生对应的易感机制和功能。
我们将在基因组学以及比较基因组学,群体遗传学综合层面上深入探索疾病基因组和癌症基因组。
实验设计与样本(1)Case-Control 对照组设计;(2)家庭成员组设计:父母-子女组(4人、3人组或多人);初级数据分析1.数据量产出:总碱基数量、Total Mapping Reads、Uniquely Mapping Reads统计,测序深度分析。
2.一致性序列组装:与参考基因组序列(Reference genome sequence)的比对分析,利用贝叶斯统计模型检测出每个碱基位点的最大可能性基因型,并组装出该个体基因组的一致序列。
3.SNP检测及在基因组中的分布:提取全基因组中所有多态性位点,结合质量值、测序深度、重复性等因素作进一步的过滤筛选,最终得到可信度高的SNP数据集。
并根据参考基因组信息对检测到的变异进行注释。
4.InDel检测及在基因组的分布: 在进行mapping的过程中,进行容gap的比对并检测可信的short InDel。
在检测过程中,gap的长度为1~5个碱基。
对于每个InDel的检测,至少需要3个Paired-End序列的支持。
5.Structure Variation检测及在基因组中的分布: 能够检测到的结构变异类型主要有:插入、缺失、复制、倒位、易位等。
技术路线图(国自然)
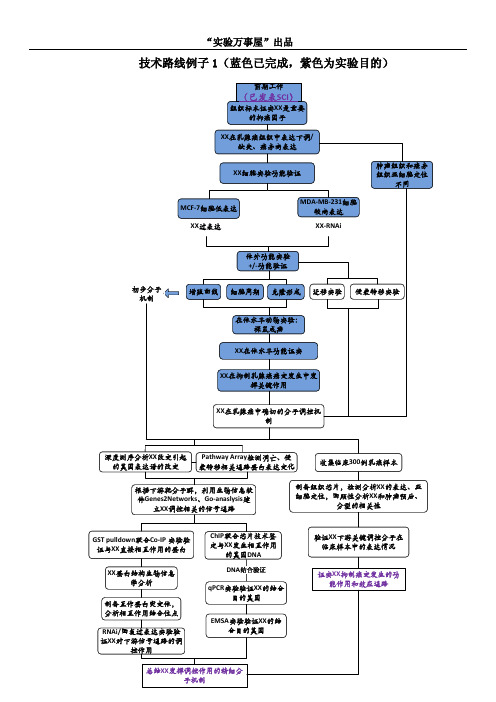
验证XX下游关键调控分子在 临床样本中的表达情况
证实XX抑制癌变发生的功 能作用和效应通路
“实验万事屋”出品
例子 2(细胞水平、组织水平分开画图,如果有动物水平再另外画一个) 1、细胞水平:
2、组织水平
“实验万事屋”出品
例子 3
“实验万事屋”出品 例子 4
DNA蛋白
Papnet 筛查宫颈癌变
蛋白DNA
初步分子 机制
体外功能实验 +/-功能验证
增殖曲线 细胞周期 克隆形成 迁移实验 侵袭转移实验
在体水平动物实验: 裸鼠成瘤
XX在体水平功能证实
XX在抑制乳腺癌癌变发生中发 挥关键作用
XX在乳腺癌中确切的分子调控机 制
深度测序分析XX改变引起 Pathway Array检测凋亡、侵
的基因表达谱的改变
袭转移相关通路蛋白表达变化
HPV致病DNA正向示踪分析
HPV致病蛋白逆向求源分析
分离克隆宫颈癌细 胞的表达差异DNA
核酸印迹杂交分析 筛查致病DNA
合成相应mRNA 生物探针
表达谱基因芯片 杂交分析
鉴定相应蛋白 的差异表达
病毒致癌分子 调控机理分析
HLA系统
P53、Rb、 E2F-1
端粒体系统
细胞素、 活化因子
致癌相关蛋白 一级分析
XX蛋白结构生物信息 学分析
制备互作蛋白突变体, 分析相互作用结合位点
RNAi/回复过表达实验验 证XX对下游信号通路的调
控作用
ChIP联合芯片技术鉴 定与XX发生相互作用
的基因DNA
DNA结合验证
qPCR实验验证XX的结合 目的基因
EMSA实验验证XX的结 合目的基因
总结XX发挥调控作用的精细分 子机制
乳腺癌基因组学研究进展

乳腺癌基因组学研究进展张柏林;宋丰举【摘要】乳腺癌的发生是由体细胞突变引起。
研究乳腺癌的体细胞突变谱有助于明确乳腺癌发生发展的生物学过程。
应用二代测序技术对乳腺癌基因组学的研究有了一系列新的认识。
二代测序技术检测到新的乳腺癌相关基因,这些基因的突变频率较低,不同患者的突变基因却涉及某些通路的失调。
某些乳腺癌基因组中可识别特异性突变签名,但一般不反映环境暴露。
尽管所有肿瘤中瘤内异质性均存在亚克隆突变,均有一个优势克隆占全部乳腺癌细胞的50%以上。
乳腺癌基因组学旨在促进向个体化医学转化,基于基因组信息的乳腺癌分子分型和个体化治疗将在不远的将来成为现实。
%Breast cancer is caused by somatic mutation. As such, somatic mutation in breast cancer should be described to eluci-date the underlying mechanism. Next-generation sequencing has provided new insights into the genomics of breast cancer. New genes were identified and exhibited a relationship with breast cancer. Although these genes mutated at a low frequency, such genes in different cases could be categorized into specific pathways. Mutational signatures could be found in some cases, but such signatures were gener-ally not related to environmental exposure. Studies on intra-tumoral heterogeneity have revealed the ubiquitous presence of sub-clones in breast cancer;however, a major clone is also observed, accounting for>50%of tumor cells. Current advancements show that breast cancer genomics has been integrated into personalized medicine. Furthermore, a genome-informed and personalizedmolecular sub-typ-ing and treatment of breast cancer can be developed in the future.【期刊名称】《中国肿瘤临床》【年(卷),期】2014(000)003【总页数】4页(P207-210)【关键词】乳腺癌;基因组学;二代测序;个体化医学;体细胞突变【作者】张柏林;宋丰举【作者单位】天津医科大学肿瘤医院放疗科,国家肿瘤临床医学研究中心,天津市肿瘤防治重点实验室,乳腺癌防治教育部重点实验室天津市300060;天津医科大学肿瘤医院流行病室,国家肿瘤临床医学研究中心,天津市肿瘤防治重点实验室,乳腺癌防治教育部重点实验室天津市300060【正文语种】中文乳腺癌是全球女性发病和死亡占第1位的恶性肿瘤[1]。
全基因组扫描在抗癌基因筛选中的研究

全基因组扫描在抗癌基因筛选中的研究随着肿瘤学的快速发展以及基因测序技术的日益完善,现代医学已经进入了基于基因的个性化抗癌治疗时代。
而全基因组扫描就是其中一个重要的手段之一。
本文主要分为以下几个方面来探讨全基因组扫描在抗癌基因筛选中的研究。
一、什么是全基因组扫描全基因组扫描是指对人类全基因组进行测序,并对其进行全面的基因组分析的方法。
这种技术主要分为两种方法:第一种是短读长度测序技术,主要是指Illumina、SOLiD、454等;第二种为长读长度测序技术,例如PacBio和Oxford Nanopore等。
目前大多数全基因组测序都采用了第一种方法。
这种方法可以在较短的时间内测序整个基因组,并产生大量的序列数据。
二、全基因组扫描在癌症基因筛选中的应用现在,全基因组扫描已成为癌症基因筛选和新型抗癌药物开发的重要手段。
因为随着人们对基因的研究逐渐深入,越来越多的人发现癌症是以基因突变为基础的。
全基因组扫描是一种技术和方法,可以帮助医学工作者更全面地理解人类基因组,从而推动下一步基于基因的治疗手段的开发。
三、全基因组扫描的应用流程1.样本采集和总DNA提取2.质控3.高通量测序4.数据处理和分析全基因组扫描应用流程中一般会先对样本进行采集和总DNA 提取。
在这一步中,需要从人体中获取DNA样本,并将其提取至高纯度的样本。
接下来就是质控环节。
质控环节主要用来除去一些低质量的样本来确保后续流程的准确性和可靠性,一般包括三方面:1.测序质量控制:光学密度和光碟恒温度。
2.检测序列的碱基质量3.剔除掉low quality的reads经过了质控部分,就可以正式进入到高通量测序环节。
全基因组扫描通常采用高通量测序技术,如Illumina和SOLiD等。
最后,数据处理和分析是流程中的一个重要环节。
数据处理和分析主要包括两个方面,一个是基因组序列的比对,一个是突变筛选。
最后确定是否选择这个基因作为我们后续抗癌基因的筛选对象。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
Landscape of somatic mutations in 560 breast cancer whole
genome sequences
Nature. 2016 Jun 2;534(7605):47-54. doi: 10.1038/nature17676
IF: 38.138
背景
1.癌症的基因突变理论指出“司机”(driver)突变使肿瘤细胞获得增殖优势,随之发生的“乘客”(passenger)也与肿瘤发生相关。
这些突变主要来自于内源性或外源性诱变剂暴露、异常DNA 编辑、复制错误和DNA修复缺陷;
2.DNA测序技术可系统的分析肿瘤基因的各类突变,包括单碱基替换、小的插入/缺失、重排及拷贝数变异;
3.目前大多乳腺癌基因组研究集中在蛋白编码的外显子区域,对非翻译区、内含子和基因间区研究有限,对揭示乳腺癌分子致病机制具有局限性。
研究方法
1. 样本收集
●560例乳腺癌患者外周血淋巴细胞样品DNA
●560例病灶相邻正常乳腺组织或皮肤样品DNA
●268乳腺癌患者totalRNA
2. 测序及比对
●测序平台:Illumina GAIIx, Hiseq 2000 或Hiseq 2500
●肿瘤样品平均测序深度40.4X,正常对照样品平均测序深度30.2X
3. 基因组数据处理
4. 鉴定新的乳腺癌基因
5. 非编码区域常见变异分析
6. 变异特征分析
7. Kataegis突变分析
8. 重排突变分析
●分类:聚簇类重排,非聚簇类重排
●亚分类:缺失、倒位、串联重复, 1-10kb、10kb-100kb、100kb-1Mb、
1Mb-10Mb、>10Mb(染色体间易位)
研究结果——肿瘤基因及driver突变
560例乳腺癌患者亚型分类
●共检测到3,479,652个体细胞单碱基变异、371,993 个小的插入/缺失变异、77,695 个重排突变;
●结合其他研究,共分析了1,332个乳腺癌患者的测序数据,完成了包含727个肿瘤基因的清单(包括MED23、FOXP1、MLLT4、XBP1和ZFP36L1共5个之前罕有研究或不明确的基因)。
图为93个Driver基因突变在ER阳性或阴性患者体内分布 检测出1,278个特有的、39个罕见的基因阅读框内融合。
对260例患者转录组测序分析,未发现这些融合基因的表达。
影响CDKN2A、RB1、MAP3K1、PTEN、MAP2K4、ARID1B、FBXW7、MLLT4 和TP53基因印记的常见重排突变远高于重排率背景。
检出显性效应基因ETV6和ESR1部分区域频繁发生重排
突变,但转录组水平无变化,突变意义未知。
检测出93个基因的1,628个疑似driver突变,95%的患者至少检测到1个driver突变,Top10突变基因为TP53、PIK3CA、MYC、CCND1、PTEN、ERBB2、chr8:ZNF703/FGFR1 位置、GATA3、RB1 和MAP3K1。
Top10基因突变占疑似driver突变的62%。
driver突变各种类型占比
研究结果——非编码区域的常见体细胞变异
图为5个频繁发生突变的调控区域的单碱基替换或Indel变异分布,右图为分别包含这5个区域突变的样品及560个样品总的12类变异特征分布比例
●检测到5个频繁发生突变的调控区域,PLEKHS1基因启动子区域CAGCAAGC TGAACA GCTTGCTG回文重复序列之间的G和C位置、TBC1D12启动子Kozak区域CCCCAGATGGTGGG的C和G位置、WDR74基因启动子区域、长链非编码RNA MALAT1和NEAT1
研究结果——变异特征
●检测到12类单碱基特征变异,特征5, 6, 17, 18 和20之前在其他癌种类型中检测到,特征26和30为新检测到的类型;
●结合患者的生物学特性,特征1和5与诊断年龄相关、特征2和13和APOBEC相关、特征6, 20和26与MMR缺陷相关、特征3和8和HR缺陷相关、特征18, 17 和30 为病因不明类型。
上图显示在每个样品中每类特征变异的数量
下图显示在每个样品中每类特征变异的比例
含有12类特征变异的样品比例及样品中每类特征变异的数量分布
●检测到6类特征变异,特征1突变(占重排突变的9%)和特征3突变(占重排突变的18%)主要特点是具有串联重复,特征1突变大多>100kb,特征3突变<10kb。
>95%的特征3串联重复突变在15%的癌种中检测到,91%的BRCA1突变或启动子区域甲基化均属于特征3突变。
>35%的特征1串联重复突变仅在8.5%的乳腺癌患者中检测到,具有该类突变的患者通常有TP53突变,但无BRCA1/2突变或BRCA1启动子区域甲基化。
●特征5突变(占重排突变的14%)的特点是具有<100kb的缺失,常与BRCA1突变或启动子区域甲基化、BRCA2突变及特征1的大片段串联重复突变同时发生。
●特征2突变(占重排突变的22%)的特点是具有非聚簇类>100kb的缺失、倒位和染色体间易位,该类突变在ER阳性患者中较常见。
●特征4突变(占重排突变的18%)的特点是染色体内聚簇类染色体易位。
●特征6突变(占重排突变的19%)的特点是聚簇类倒位和缺失。
miRNA聚簇分析结果,0=红色, 1=紫色, 2=蓝色, 3=浅蓝色, 4=绿色, 5=橙色
BRCA状态,BRCA1突变显示为紫色,BRCA2显示为橙色
AIMS亚型,(basal=红色, luminal B=浅蓝色, luminal A=深蓝色)
GISTIC聚簇分析结果,MYC、ZNF217、ZNF703、CCND1及ERBB2扩增情况,同源重组缺陷评分(HRD),Kataegis突变分布,GATA3、PIK3CA、PTEN、RB1及TP53基因突变分布,HER2、PR及ER状态,黑色显示为阳性
结合重排对每类变异特征贡献率、组织学、基因表达将6类重排特征变异划分为7个亚类
上图显示为7个亚型变异样本的6类重排特征变异分布比例下图显示为7个亚型变异样本中12类单碱基变异特征分布
携带7个亚型特征变异样本的OS情况
携带7个亚型特征变异样本的确诊年龄、肿瘤分级和绝经状态
携带7个亚型特征变异样本的ER状态、免疫响应及淋巴细胞浸润评分
局部突变簇(kataegis )在49%的乳腺癌患者中检出,通常和具有聚簇类重排的特征4和6突变同时发生,而具有串联重复或缺失的特征1、3和5突变通常无kataegis突变
总结
√ 整个染色体拷贝数变异及非目标区域的常见突变可能包含其他肿瘤相关基因;
√ 非编码区域的driver突变鉴定需要更多测序研究;
√ 病毒或其他微生物在肿瘤发生过程中的作用有待进一步研究;
√ 完全理解肿瘤的体细胞突变基础需要更多的全基因组测序研究。