化合物价电子结构及晶体结合能计算
2.2 晶体的结合类型

2.3.5 氢键结合与氢键晶体
• 氢原子的电子参 与形成共价键 后,裸露的氢核 (质子)与另一 电负性较大的原 子通过静电作用 相互结合。
机动
目录
上页
下页
返回
结束
1. 由上讨论可知,原子结合成晶体时,是以以上哪种结合力 结合,很大程度上决定于它电负性特性。 2. Ⅴ、Ⅵ、Ⅶ族元素也可以形成共价键,但由于共价键的饱 和性,Ⅴ族元素只能形成三个共价键,Ⅵ、 Ⅶ 族元素则 只能分别形成二个,一个共价键,但仅有三个、二个、一 个共价键不能形成三维晶体。所以对Ⅴ族元素三个共价键 常在一个平面上,形成层状结构,而各原子间则靠范德瓦 尔斯力结合,对Ⅵ族元素,二个共价键常形成环状结构, 各个环之间依靠范德瓦尔斯力结合;对于Ⅶ族元素,常由 一个共价键先组成分子,而分子之间依靠范德瓦尔斯力形 成分子晶体。 3. 实际晶体的结合往往不是纯属哪一种键,而是包含两种或 更多种键,任何晶体都包含范德瓦尔斯键(电子分布的起 伏而产生的瞬时偶极矩总存在)。
—— 良好的导电本领 —— 结合能比前面两种晶 体要低一些 —— 过渡金属结合能较大
机动
目录
上页
下页
返回
结束
晶体的平衡 —— 依靠库仑作用力和一定的排斥力 排斥来自两个方面 —— 体积减小,电子云的密度增大,电子的动能将增加 —— 原子实相互接近到一定的距离时,它们的电子云发生 显著的重叠,将产生强烈的排斥作用 —— 金属性结合对原子的排列没有特殊的要求,容易造成 原子排列的不规范性,使其具有很大的范性
机动 目录 上页 下页 返回 结束
共价键结合的两个基本特征 —— 饱和性和方向性 饱和性 —— 共价键结合的原子能形成键的数目有一个最 大值,每个键有2个电子,分别来自两个原子 —— 共价键是由未配对的电子形成 —— 价电子壳层如果不到半满,所有电子都可以是不配 对的,因此成键的数目就是价电子数目 —— 价电子壳层超过半满时,根据泡利原理,部分电子 必须自旋相反配对,形成共价键数目小于价电子数目 IV族 — VII族的元素共价键数目符合8-N原则
晶体化学基本原理、结构与关系

离子键分数与电负性差值(XA-XB)的关系
电负性差值越大,离子键分数越高。
晶体化学基本原理、结构和关系
当两个成键原子的电负性相差很大时,如周期表 中I-VII 族元素组成的化合物,主要是离子键;
电负性相差小的元素的原子之间成键,主要是共 价键,也有一定的离子键成份,价电子不仅为两 原子共享,而且应偏向于电负性大的原子一边;
特 点:无方向性和饱和性
晶体化学基本原理、结构和关系
晶体化学基本原理、结构和关系
电子云
金属原子
金属晶体特征:配位数较高、密度大、电阻随温度
升高而增 大、 强韧性好、导电和导
热性良好、特有金属光泽
晶体化学基本原理、结构和关系
(4)分子间力(范德华键——I2)
正负电荷中心不重合
极化原子之
本 质:原子(分子、原子团)之间的 间吸引力 偶极矩作用 —— 分子间力
共价键 金属键
范德华键
物理键
(派生结合 或二次键)
氢键 离子极化 ……
晶体化学基本原理、结构和关系
(1)离子键(NaCl)
本 质:正负离子之间的静电吸引作用 特 点:结合力大、无方向性和饱和性
晶体化学基本原理、结构和关系
晶体化学基本原理、结构和关系
离子晶体特征:配位数较高、硬度高、强度大、熔 点较高、常温绝缘、熔融后导电、 无色透明.
无
共价键 63-712
高
高
不导电
有
金属键 113-350
有高 有低
有高 有低
良好
无
分子键 <42
低
低
不导电
有
晶体化学基本原理、结构和关系
晶体化学基本原理、结构和关系
xps中化合价和结合能之间的关系

XPS是X射线光电子能谱的简称,它是一种常用的表面分析技术。
在实际应用中,XPS能够提供物质的化学状态、元素的化合价以及元素之间的结合能等相关信息。
而化合价和结合能是物质的重要性质,它们之间的关系对材料科学和表面化学的研究具有重要意义。
化合价是指化学元素在化合物中所呈现的电价状态。
化合价的大小对物质的化学性质起着重要影响。
而XPS技术通过对物质表面原子的电子结构和分布进行测定,可以辨别化合物中元素的化合价状态。
这主要是通过X射线照射原子表面,使之发生电子光电子发射,根据光电子的动能进行分析,得到元素的电子结合情况,从而推断出元素的化合价。
结合能是指在分子或晶体中最紧密的两个原子核之间的相互作用能,它是描述分子内相对原子位置的势能。
结合能直接反映了原子之间的相互作用程度和原子在分子中的稳定性。
XPS技术通过对物质的电子能谱进行测定,可以计算得到原子的结合能。
这是因为XPS技术能够测定物质中原子的电子分布和能级情况,从而推算出原子之间的相互作用能。
化合价和结合能之间的关系是密切相关的。
物质中元素的化合价状态直接影响了原子的电子结构和能级分布,从而影响了XPS谱峰的位置和能量分布。
另物质中原子之间的结合能与化合价状态密切相关,原子的结合能大小直接决定了化合物的稳定性和化学性质。
通过XPS技术测定化合物中元素的化合价和结合能,可以全面了解物质的化学性质和结构特征。
在实际应用中,XPS技术通过分析化合物的表面电子能谱,可以得到元素的化合价状态以及原子间的结合能。
这对于材料科学和表面化学的研究具有重要意义。
在材料表面的功能化改性过程中,XPS技术可以分析表面原子的化合价状态变化以及化合物与表面基底的结合能变化,从而揭示材料表面化学反应的机理和规律。
又如,在催化剂的研究中,XPS技术可以通过分析催化剂表面原子的化合价状态和原子间的结合能,探究催化剂的表面活性位点和反应活性的关系。
XPS技术通过分析化合物的表面电子能谱,可以得到物质中元素的化合价状态和原子间的结合能。
Au-Cu系金属间化合物价电子结构及晶体结合能计算

能 。 计 算 结 果 表 明 :金 属 问化 合 物 A u uC 、Au u u u 的最 强 键 分 别 为 Au A 键 、A A “ 、Au C C 、A C 3 ~ u um u键 — u
键, 强键键 能分别为 1 . 86 l.3 和 1.6 J l晶体理 论结 合能分别为 4 1 5 3 36 最 07 、 0 8 2 0 O130k/ , mo 0 . 、6 .4和 3 1 2 J l 2 8. / 。 0 k mo 用 E T理论计算的晶体结合能值与用特征 晶体理 论计 算的晶体结合能值基本吻合 。3种 化合 物中 ,A u的最强 E uC
cl l e ae ntee ic l l t nte r f oisadmoeue E T . h eut so ta tes o gs a ua db sdo h mpr a e c o hoy o l n l l c t i er s d c s(E ) T ersl h w th t n et s h r
b n so 3 u Au n Cu r — Au b n , m Au I o d a d Au C o d r s e t ey t ee e g e f o d f Au C , Cu a d Au 3 eAu a o d Au I 1b n n — u b n e p c i l , h n r iso v
键 键 能 和 品 体 理 论 结 合 能 最 大 , 因此 其 稳 定性 最好 。 关 键 词 :AuC — u体 系 ; 金 属 问 化 合 物 ;经 验 电子 理 论 ;价 电 子 结 构 ; 结 合 能 中图分类号:O 4. 6 11 文 献 标 志 码 :A
C l u a i n o a e c l c r n s r t e nd c he i e e r i so a c l to f l n ee e t o tuc ur sa o sv ne g e f v
第二章 晶体的结合3、4(共价结合)详解

- V22 V32 - V3
V2
15
结论:当原子A和原子B为异类原子时,所形成的共 价键包含有离子成份,所形成的共价键在两原子间是 不均衡的,或这种情况下,采取的是共价结合与离子 结合之间的过渡形式。
四、有效离子电荷
对Ⅲ-Ⅴ族化合物,以GaAs为例子来说明
离子实分别带+3q与+5q的离子Ga3+和As5+,而每一 对Ga与As有8个电子。
2)电子处于杂化轨道上,能量比基态提高了(形 成杂化轨道需要一定能量),但杂化后,成键数目增 多了,且由于电子云更加密集在四面体顶角方向,使 得成键能力更强了,形成共价键时能量的下降足以补 偿轨道杂化的能量。
11
12
三、不同种原子形成的共价键
VA VB
A B
仍采用分子轨道法,约化为单电子方程
满足薛定谔方程当原子相互靠近波函数交叠形成共价键个电子为两个氢原子所共有原子核的库仑势描写其状态的哈密顿量seq下标a和b代表两个原子1和2代表两个电子分子轨道法忽略两个电子之间的相互作用v12简化为单电子问题假定两个电子总的波函数满足seq单电子波动方程分子轨道波函数两个等价的原子a和b归一化常数c取分子轨道波函数为原子轨道波函数的线性组合两种分子轨道之间能量差别1负电子云与原子核之间的库仑作用成键态能量相对于原子能级降低了反键态的能量升高
PA
1
1 2
, PB
2 1 2
Ⅲ族原子(即B原子,如Ga原子)的有效电荷数为
qB
*
(3
8
1
2 2
)
Ⅴ族原子(即A原子,如As原子)的有效电荷数为:
q
A
*
(5
8
1
1
晶体结合能的定义

晶体结合能的定义
晶体结合能是指晶体中各个原子或离子之间相互结合形成晶格时释放出的能量。
在晶体中,每个原子或者离子都受到周围其他原子或离子吸引或排斥的力,这种相互作用力决定了晶体的结构和稳定性。
晶体结合能的大小直接影响着晶体的熔化点、硬度、脆性等性质。
晶体结合能的计算是固体物理中的重要课题之一。
从相互作用的角度来看,晶
体结合能可以分为键结合能和晶胞内能。
键结合能是指相邻原子之间由于共价键、离子键或金属键形成的结合能,它决定了晶体的化学性质。
晶胞内能则是指晶格中所有原子或离子的相互作用能量总和,它反映了晶格的结构稳定程度。
在考虑晶体结合能时,不仅需要考虑相邻原子之间的相互作用,还需要考虑晶格中不同原子或离子之间的相互作用以及晶体构造的对称性。
晶体结合能的计算方法有多种,常见的包括经验势函数方法、第一性原理计算
方法等。
在实际应用中,晶体结合能的计算可以帮助科研人员理解晶体的性质、设计新材料,以及预测晶体的稳定性和相变行为。
随着计算力学和材料科学的发展,晶体结合能的计算方法越来越准确,为材料设计和应用提供了重要的理论支持。
总之,晶体结合能是描述晶体中原子或离子之间相互作用的重要物理量,它反
映了晶体的结构稳定性和各种物理性质。
通过计算和研究晶体结合能,可以深入了解晶体的性质,为材料科学和工程技术提供重要参考。
以上是关于晶体结合能的定义和相关内容,希望可以对读者有所帮助。
第二章晶体结构结合力和结合能
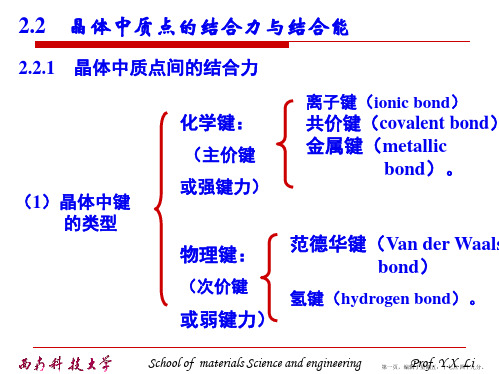
较弱 无方向性键、结构密堆、低 熔点、绝缘
氢键 氢原子核与极性分子 弱 有方向性和饱和性 间的库仑引力School of materials Science and engineering Prof. Y.X. Li 第五页,编辑于星期五:十七点 四十九分。
2.3 晶体中质点的堆积
2.3.1 原子半径和离子半径 原子半径或离子半径
有效半径
离子半径 共价半径 金属半径
一种原子在不同的晶体中,与不同的元素相结合时, 其半径有可能发生变化。晶体极化、共价键的增强和配位 数的降低都可使原子或离子之间距离缩短,而使其半径减 小。
School of materials Science and engineering Prof. Y.X. Li 第十三页,编辑于星期五:十七点 四十九分。
对于1molAX型晶体,原子总数N=2N0,于是晶格
能计算如下:
EL
N0 Az1z2e2 1 1
4 0r0
n
取决于晶体 结构类型
式中:A-马德龙常数见表2.4(P24)
n-Born Index.(与离子的电子层结构有关)。
ε0-真空的介电常数,
取决于电子 ≈8.85×10-12C2.N-1m-2 层结构
School of materials Science and engineering Prof. Y.X. Li 第二十一页,编辑于星期五:十七点 四十九分。
2.3.3 内在因素对晶体结构的影响-化学组成 (1)原子或离子半径
•原子半径或离子半径是定值吗?
•原子半径或离子半径的大小与哪些因 素有关?
氢键:指氢原子同时与两个电负性很大而原子半径较
第二章 晶体结合

方向性------各个共价键之间有确定的取向。 成键时,电子云发生交叠,交叠越多键能越大,系统 能量越低,键越牢固。
例如:金刚石结构的4个键的方向是沿着正四面体的4 个顶角方向,键间的夹角恒为109028‘。
特性:
特性差别较大。典型的原子晶体,具有熔点高、热 膨胀系数小,导电性能差、硬度高等特点。 例如: 从熔点来看,金刚石约为3280k、而Si为1693k,Ge 为1209k。 从导电性来看,金刚石是一种良好的绝缘体,而Si 和Ge在极低温度下才是绝缘体,同时它们的电阻率 随温度升高而急速的下降,是典型的半导体材料。
结合能 强 数ev/键
稳定的正、负离子相 间排列通过库仑静电 力相互吸引。
熔点高:硬度大,膨胀系数 小,易沿解理面劈裂,高温 下有良好的离子导电性。
周期表左右两 边负电性差异 大的原子之间 形成结合。 负电性接近且 较大的原子或 同种原子相互 结合。
共价键:两原子共有 的自旋相反配对的电 子结构。
完整晶体硬度大, 熔点一般较高, 低温下导电性能较差,为绝缘体 或半导体。化学惰性大,由于饱 和性、方向性,决定了原子排列 只能取有限的几种形式。
四、电负性
度量原子吸引电子的能力。一般选定某原子的 电负性为参考值,其他原子的电负性与此参考值作 比较。
穆力肯提出的电负性定义为: 负电性=0.18(电离能+亲和能) 常数的选择以方便为原则,例如一种常用的选择方 法:为使锂(Li)的负电性为1,选上常数为0.18。
泡林提出的电负性计算为:
E(A-B)= [E(A-A)×E(B-B)]1/2+96.5(xA-xB)
xA,xB 原子A和B的电负性;
E(A-B):双原子分子AB的离解能
E(A-A) :双原子分子AA的离解能
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
Au-Cu系金属间化合物价电子结构及晶体结合能计算
作者:蒋淑英, 李世春, JIANG Shu-ying, LI Shi-chun
作者单位:中国石油大学(华东)机电工程学院,东营,257061
刊名:
中国有色金属学报
英文刊名:THE CHINESE JOURNAL OF NONFERROUS METALS
年,卷(期):2010,20(4)
1.BRANDES E A;BROOK G B Smithells metals reference book 1992
2.张瑞林固体与分子经验电子理论 1993
3.YU Fang-xin;XIE You-qing;NIE Yao-zhuang Electronic structure of Au-Cu alloys[期刊论文]-Transactions of Nonferrous Metals Society of China 2004(06)
4.SHOCKLEY W Order-Disorder transformation in Au-Cu alloy[外文期刊] 1938
5.余瑞璜固体与分子经验电子理论 1978(04)
6.CLAESON T;BOYCE J B Order-disorder transformation in Au-Cu alloys studied by extended x-ray-absorption fine structure 1984
7.OKAMOTO H;CHAKRABARTI D J;LAUGHLIN D E;MASSAlSKI T B The Au-Cu(gold-copper) system 1987(05)
8.张建民;张瑞林;余瑞磺;郑伟涛 胡安广Fe-Al合金有序无序相变的电子理论研究 1994(05)
9.李文;张瑞林金属间化合物的价电子结构脆性判据 1999(01)
10.李文;关振中;杜立明;支文 殷方信 张瑞林 余瑞璜Ti-Al系金属间化合物价电子结构对其脆性的影响 1996(11)
11.高英俊;钟夏平;刘慧;吴伟明微量Sc对Al-Mg合金晶粒细化影响的电子结构分析 2002(z2)
12.杜晓东;丁厚福;宣天鹏CrB价电子结构对其性能的影响[期刊论文]-中国有色金属学报 2005(12)
13.刘志林;孙振国;李志林余氏理论和程氏理论在合金研究中的应用 1998(02)
14.贾堤;董治中;于申军;刘文西TiMe合金的价电子结构分析及结合能计算 1998(03)
15.陈舜麟;顾强;王天民Co3Ti与CoTi的晶体结构与结合能的计算及其脆性 1995(06)
16.徐万东;张瑞林;余瑞璜过渡金属化合物晶体结合能计算 1988(03)
17.Orr R L Heats of formation of solid Au-Cu alloys 1960(07)
18.ORIANI R A Thermodynamics of liquid Ag-Au and Au-Cu alloys[外文期刊] 1954
19.TOPOR L;KLEPPA O J Thermochemistry of binary liquid gold alloys[外文期刊] 1984
20.SYKES C;EVANS H The transformation in the copper-gold alloy CU3Au 1936
本文链接:/Periodical_zgysjsxb201004024.aspx。