美国医疗器械法规体系--QSR820介绍
qsr820法规解读
820.25
人员
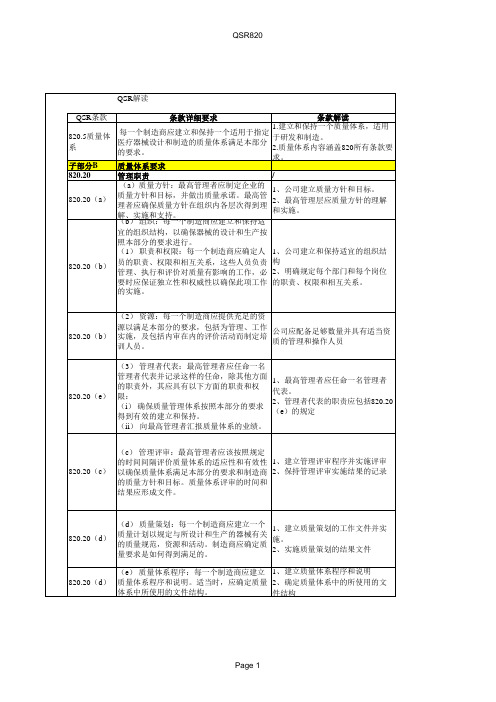
(a) 总则:每一个制造商应具有充足的人 公司应配备足够数量并具有适当资 820.25(a) 力资源,人员应具备必要的教育背景、培训 质(教育背景、培训、经验)的管 理和操作人员;审核过程进行评估 和经验以确保完成本部分所要求的活动。 (b) 培训:每一个制造商应建立一个识别 培训需求的程序以确保所有人员能够接受适 宜的培训以完成本职工作。培训应形成文件 。 1、建立人员培训的管理文件 820.25(b) (1)作为培训的一部分,工作人员应了解 2、保持人员培训的记录 由于其不恰当的操作而造成的器械缺陷。 (2)进行验证和确认活动的人员应了解在 工作中可能遇到的器械缺陷和故障。 子部分C 820.30 设计控制 设计控制
820.70(a)
(1)形成文件的指导书,标准的操作程序 建立生产过程控制的作业指导书或 (SOP’S),规定和控制生产方式的方法; 操作规程(SOP)
820.70(a)
(2)生产过程中,过程参数、组件和器械 特性的监视和测量;
建立生产过程中,过程参数、组件 和器械特性的监视和测量的作业指 导书或或操作规程(SOP) 过程控制应符合相应的参考标准或 代码 生产过程所使用的设备和过程应经 过批准
子部分D 820.40
文件控制 文件控制
/
制造商应建立和保持程序已控制所有本部分 要求的文件,程序应规定以下内容: (a) 文件的批准和发布:每一个制造商应 指定人员在文件发布前,评审文件的适应性 和批准文件,以确定满足本部分的要求。文 建立和保持文件控制程序,文件控 820.40(a) 件的批准包括批准的日期和批准人的签名应 制程序的内容至少包括820.40 形成文件。满足本部分要求的文件应在使用 (a)规定的内容 现场方便获得,或必要时获得。应及时清除 使用现场所有的作废文件,以防止非预期的 使用。
(最新)qsr820中文版本

1.1 概述§820.1 范围(a) 适用性(1) 在这个质量体系规范中描述了现行的生产管理规范的要求(CGMP). 本规范要求规定了所有医用器械成品在设计,制造,包装,标签,贮存,安装和服务中使用的方法,设施和控制.这些要求是为了确保医疗器械成品的安全和有效,并遵从美国食品,药品和化妆品法.本规范提出了适用于医疗器械成品制造商的基本要求.如果某制造商只进行本规范规定的以部分操作,而不进行其他操作,则该制造商仅需执行适用于他所进行操作的那些要求.有关Ⅰ类器械,设计控制仅按在§820.30(a)(2)中列出的要求进行.这个规范不适用于成品组件和零件的制造商,但鼓励这样的制造商使用规范的适当规定作为指导.人类血液制品和血液成分的制造商不属于本规范的管理范围,但属于606的管理范围.(2) 这一规范的规定适用于本规范定义的医疗器械成品,即使用的对象是人的,在美国各州或领地,哥伦比亚特别区或波多黎哥联邦制造,进口或出口的器械成品.(3) 在本规范中,几次使用了短语”适当的地方”. 当要求以”适当的地方”来限制时,如果制造商没有合理的理由来证明不适宜,就认为此要求是”适当的”. 如果不贯彻”适当的”要求,就会导致产品达不到要求或制造商不能采取某些必要的正确措施.(b) 范围这一的质量体系规范和增补在这一规范其他节的条文明确注明用于其他方面的条文除外. 在不可能执行全部适用条文(包括这一节的和这一规范其他节的条文)的情况下,特定的运用于有问题器械的条文将取代其他一般的适用条文.(c) 权威性根据联邦法501, 502, 510, 513, 514, 515, 518, 519, 520, 522, 701,704, 801, 803, (21U.S.C.315, 352, 360, 360c, 360d, 360e, 360h, 360i, 360j, 360l, 371, 374, 381, 383) 建立和提出了820规范的权威性. 如果没能执行适用规范,可能会导致产生伪劣器械, 根据法规501(h), 对这样的器械和未能执行规范的人都要进行处罚.(d) 外国制造商如果某制造商提供给美国的进口器械,拒绝接受FDA对外国设备进行是否执行本规范的检查,就会出现法规的801(a)节中的后果. 用这样的设备生产出的任何器械,在设计,制造,包装, 标签,贮存,安装或服务方面,使用方法,设备和控制都未遵从法规的520(f)节和本规范的要求. 根据法规501(h), 用这样的设备制造出来的器械都属于伪劣产品.(e) 豁免或更改(1) 任何希望豁免或更改执行某些器械质量体系要求的申请,都要遵从法规520(f)(2)的要求. 申请豁免或更改的过程将依据这一规范§10.30的程序进行,即FDA的管理程序. 以下地址可提供指导: 器械和放射卫生中心, 小制造商处(HFZ-220), 1350 Picca rd Dr., Rockville, MD20850, U.S.A. 电话: 1-800-638-2041或1-301-443-6597, FAX 301-443-8818.(2) 当FDA判定某种更改有益于公众健康时, 就会起草并认可这项更改. 这种更改仅能在一段时间内维持有效, 即当器械仍能满足公众健康需要, 并且如果没有更改,器械不可能制造得非常有效的一段时间内.§820.3 定义(a)法指的是美国食品,药品和化妆品法修正案(secs.201~903, 52Stat.1040 et seq.,修正版21U.S.C.321~394). 在法201中的全部定义都适用于本规定.(b)投诉指的是以某些书面的,电子的或口头的形式表达意见, 认为在配发后的器械在鉴定,质量,耐久性,可靠性,安全性,有效性或性能方面有缺陷.(c)组成指的是原材料,物质,小件,零件,软件,硬件,标签或有包装和标签的成品器械的零配件.(d)控制编号指的是有区别的符号,如字母或数字的不同组合,或以原制造,包装,标签和分发的单个或批量成品的区别符号来分辨.(e)设计历史文件(DHF)指的是描述某医疗器械成品设计过程的有关记录.(f)设计输入是指作为器械设计基础对器械的物理和特性要求.(g)设计输出是指各设计阶段的设计成果和最终的总设计成果. 完成设计输出包括器械,包装和标签,器械主记录.(h)设计评审是指依照依照文件进行广泛,系统的设计评审, 以评价设计要求的适当性,并评价设计达到这些要求的能力,查明问题所在.(i)器械历史记录(DHR)是指医疗器械成品制造过程的记录.(j)器械主记录(DMR)是指包括医疗器械成品的程序和规范的完满记录.(k)建立是指定义,文件(书面的或电子的)和执行情况.(l)器械成品是指适于使用或具有功能的器械或器械附件, 不论是否经过包装,贴标签或灭菌.(m)批是指一种或几种组成或成品器械具有单一类型,型号,类别,尺寸,成分或软件版本,必须在相同条件下制造,并在规定的限度内具有相同的特征和质量.(n)管理职责是指制造商的高级雇员有权建立或改变制造商的质量方针和质量体系.(o)制造商是指设计,制造,构造,装配或加工成品器械的人. 制造商包括但不局限于那些从事灭菌,安装,再贴标签,再制造,再包装或Specification开发商和从事这些工作的外国实体的最初代理人.(p)生产过程副产物是指促进生产过程所用的材料或物质,制造加工过程中的伴随组分或副产品,以残余物或混杂物的形式存在.(q)不合格是指未达到特定的要求.(r)产品是指组成,制造材料,加工过程中器械,成品器械及返回器械.(s)质量是指使器械安全适用的总性质和特征,包括安全性和性能.(t)质量审核是指在规定的时间间隔,以足够的次数,对制造商质量体系进行有组织的自主的检查,检验质量体系行为和结果是否执行质量体系程序,以保证有效地执行程序,达到质量体系目标.(u)质量方针是指有关质量的机构方向和目标,是由负责的管理人员建立的.(v)质量体系是指检查质量管理的组织机构,职责,程序,处理和资源.(w)再加工是指对成品器械进行加工,调节,革新,再包装,再贮存,大大改变了成品器械的性能,安全性规范或用途.(x)返工指的是对不合格产品采取某些措施,以使他在获准配发之前达到指定的DMR要求.(y)规范是指生产,加工,服务或其他行为必须遵守的一些要求.(z)有效性是指通过检查和提供客观证据来证明能始终满足特定的用途.(1)过程确认是指通过客观的证据证明加工生产出的产物或产品始终达到预定的规范.(2)设计确认是指通过客观的证据证明器械规范与使用者的需要和设计的用途相一致.(aa)验证是指通过检查和提供客观证据来证明已经满足指定的要求.§820.5 质量体系各制造商应建立并保持一个质量体系,适合于他们设计或制造的医疗器械,并且达到本规范的要求.1.2 质量体系要求§820.20 管理职责(a)质量方针管理职能机构应建立质量方针目标和质量承诺,并保证质量方针在企业各级人员中的理解,贯彻和持续执行.(b)管理机构各制造商都应建立并维持一个适当的组织机构,以保证器械依照本规范进行设计和生产.(1)职责和权限各制造商都应任命有相应职责,权限和能独立行使职权的人员负责管理,执行和评价质量体系.(2)人员各制造商都应具备足够的合格人员,包括分派培训有素的人员从事管理,执行,评价和内部质量审核等工作,以达到本规范要求.(3)管理者代表管理职能机构应任命其中一员为管理者代表,并在文件中注明.管理者代表不论其他职责如何,必须履行下列职责和权力:i. 确保按本规范要求有效地建立和保持.ii. 向管理机构汇报质量体系进行情况,供其讨论.(c)管理评审管理职能机构应按照建立的程序,以足够的次数定期评审质量体系的适用性和有效性. 以保证质量体系达到本规范要求和制造商建立的质量方针和目标,评审日期和结果应形成文件.(d)质量策划各制造商应编制质量计划,确定与设计和制造的器械相关的质量实践,人员和措施,并建立达到质量要求的规划.(e)质量体系程序各制造商应建立质量体系的各种程序和实施指南,并形成文件.§820.22 质量审核各制造商应建立质量审核的程序,并进行管理,以保证质量体系符合建立的质量体系要求,确定该质量体系的有效性.质量审核应由与审核事物无直接责任的人执行.若有必要时,应采取措施纠正错误措施,包括对有缺陷的事物进行再审核.管理机构对各质量审核的结果及再审核的情况进行复核.提供质量审核日期和结果及再审核的有关文件.§820.25 全体工作人员(a)一般要求各制造商都应具有足够的工作人员,具备必需教育,背景,接受过培训并富有经验,以保证正确履行本节所要求的全部工作.(b)培训各制造商应建立必需培训的程序,保证全部工作人员在经过培训后能胜任他们各自的职责,并提供与培训有关的文件.(1)培训内容还包括使全体工作人员懂得由于错误执行指定工作可能会导致器械产生缺陷.(2)使从事验证和确认工作的全体工作人员能预见可能会发生的缺陷和错误. 1.3 设计控制§820.30 设计控制(a) 总则(1) Ⅱ,III类器械的制造商,以及在本规范(a)(2)段列出的Ⅰ类器械制造商,应建立并保持控制器械设计的方法,以保证达到特定的要求.(2) 下列Ⅰ类器械也需要设计控制.i.计算机软件的自动化机械.ii.下面列出的器械:(b)设计和开发计划各制造商应建立并保持有关设计和开发行为的计划,并规定执行职责.计划应规定提供或输入设计和开发程序的不同组或行为的互换信息.对计划应进行检查,用现代化手段处理并证实设计和开发的进展.(c)设计输入各制造商应建立并保持关于保证器械的设计要求适当的程序,以器械用途为主,包括使用者和病人的需要.该程序应包括关于不完善,不清楚或抵触要求的处理办法.设计输入要求应记录在文件中,并由指定的人进行检查和认可,并提供认可这些要求的日期和个人签名的文件.(d)设计输出各制造商应建立并保持关于确定和提供设计输出文件的程序,并进行执行设计输入要求的适当评价.设计输出程序应包含或制定参照的认可标准,并保证那些设计输出是鉴定器械良好性能所必需的.设计输出应记录在文件中,在获准之前进行评审,并提供有关评审认可日期和签名的文件.(e)设计评审各制造商应建立并保持一套程序, 保证在器械设计开发的适当阶段,按计划评审设计结果,并提供正式文件.评审参加者应包括设计的专业人员对设计阶段负有责任的代表和与设计阶段五直接责任的人和必要的专家.设计评审的结果包括设计鉴定,评审人员和日期,都应记录在设计历史文件(DHF)中.(f)设计验证各制造商应建立并保持验证器械设计的程序.设计验证应证明设计输出达到设计输入要求.设计验证的结果,包括设计方法的鉴定,验证人员和日期,都应当记录在DHF文件中.(g)设计确认各制造商应建立并保持设计确认的程序.应在规定的操作条件下,对试制的单个,批量产品或等同物进行设计确认的确认.设计确认应保证器械满足使用者的需要,并具有预期用途,还应包括产品在实际或设想使用条件下的试验.设计确认还应包括软件确认及适当的时候的风险分析.有关设计确认的结果,包括对设计和设计方法的鉴定,执行人员和日期都应记录在DHF文件中.(h)设计转换各制造商应建立并保持一套程序以确保器械设计正确性体现在一定的生产规范中.(i)设计更改各制造商应建立并保持一套程序,对更改的设计在执行之前进行鉴定,提供有效性文件或适当的地方进行验证,评审和认可.(j)设计历史文件各制造商应建立并保持各种类型器械的DHF. DHF应包含或参照必要的原始记录,来证明设计开发过程与认可的设计计划一致,并遵守本规范要求.1.4 文件控制§820.40 文件控制各制造商应建立并保持本规范所要求的全部文件控制的程序.程序应提供下列内容:(a)文件认可和发布各制造商应在分发达到本规范要求的全部文件之前,委派专人检查适用性和认可情况.应提供有关认可文件的日期和个人签名的文件.达到本规范要求的文件适用于指定的,使用的或其他需要的地方,所有失效的文件应从使用条款中删除.(b)文件更改更改文件应由执行原文件检查和认可的同一职能部门内的人进行检查和认可,除非有另外明确指定人选. 认可的改动应及时地转达给有关人员.各制造商应保留更改文件的记录. 更改记录应包括修改内容,相关文件的鉴定,认可人的签名,认可日期及更改生效的日期.1.5 采购控制§820.50 采购控制各制造商应建立并保持确保所有购买的或收到的产品和服务符合指定要求的程序.(a)对供应商,承包商和咨询机构的评审各制造商应建立一套供应商,承包商和咨询机构必须达到的指定要求.各制造商应:(1)根据指定要求(包括质量要求),评价和选择潜在的供应商,承包商和咨询机构.评价应记录在文件中.(2)根据评价结果,确定对产品,服务,供应商,承包商和咨询机构实施控制的方式和程度.(3)建立和保持可接受的供应商,承包商和咨询机构的记录.(b)采购资料各制造商应对采购的或收到的产品和服务建立并保留关于是否达到质量要求的资料.可能的话,应包括一份协议,关于供应商,承包商和咨询机构同意告知制造商,他们的产品或服务的改变,是制造商可以判断这些改变是否会影响成品器械的质量.采购资料应依照§820.40得到认可.1.6 标识和可追溯性§820.60 标识各制造商为防止混乱应建立并保持在接收,制造,交付和安装各阶段的产品标识程序.§820.65 可追溯性对于生产外科植入人体,支持或维持生命的器械制造商和依照制造商提供的使用说明正确使用时,如果器械运行失败可对使用者造成严重伤害,则应建立并保持对每个或每批产品都有唯一性标识的程序.程序应促进纠正错误措施.这种标识应包括在设计历史文件中.1.7 生产和过程控制§820.70 生产和过程控制(a)总则各制造商应制定,实施,控制并监测生产过程,以保证器械遵守本规范. 在制造加工过程中可能会发生违反规范的地方,制造商应建立并保持必须的生产过程控制的程序,生产过程控制应包括:(1)提供指导文件,标准操作程序(SOP’s),限定方法和生产控制方式;(2)在生产过程中监测和控制加工参数和产品特征;(3)应遵守的指定参考标准或编号;(4)加工和加工设备的认可;(5)工艺要求应阐述在工艺文件中或用通过鉴定和认可的代表性样品来表现.(b)生产和过程的改变各制造商应建立并保持改变规则,方法,加工或步骤的程序.这些改变在执行之前应被验证或在适当时依照§820.75使改变有效,这些行为均应记录在文件中.改变应依照§820.40得到认可.(c)环境控制在有理由认为周围环境条件对产品质量有不利影响时,制造商应建立并保持适当控制环境条件的程序.应定期检查环境控制系统,以核实该系统,包括必需设备的适当性,并正发挥着良好作用.检查应记录在文件中.(d)工作人员如果有理由认为工作人员和产品或环境的接触对产品质量有不利影响时,各制造商应建立并保持对工作人员的健康,卫生习惯,行为和衣着的要求. 各制造商应保证在指定的环境下临时工作的其他人员接受适当的训练或由接受过训练的人进行监督.(e)污染控制各制造商应建立并保持防止对产品质量有不良影响的物质污染设备或产品的程序.(f)厂房应该设计适当厂房,具有足够的空间进行必须的操作,以防止混乱,并保证有序的操作.(g)设备各制造商应保证在制造加工过程中使用的全部设备都达到指定要求,并经过适当设计,建造,放置和安装以利于保养,调试,清洁和使用.(1)保养计划表各制造商应建立并保持调试,清洁和其他设备保养的计划表,以保证达到生产规范.保养行为,包括执行保养行为的日期和人员应记录在文件中.(2)检查各制造商应依照建立的程序进行定期检查,以保证完成设备保养计划.检查日期和执行人员应记录在文件中.(3)调试各制造商应将设备调整限度和允许公差的说明放在需要定期调试的设备商(或附近),或者从事这些调试的工作人员都备有说明.(h)加工过程的副产物在有理由认为某加工过程的副产物对产品质量具有不利影响的情况下,各制造商应建立并保持使用和排除这种副产物的程序,以保证他被排除或减少到不会对产品质量有不利影响的量.排除或减少加工过程的副产物均应记录在在文件中.(i)自动化处理对于生产或质量体系所用的计算机或自动化数据处理系统,制造商应依照已签订的协议书验证计算机软件是否具有预想的用途.修改的软件验证有效后方能批准和发布.验证过程和结果应记录在文件中.§820.72 检验,测量和实验设备(a)检验,测量和实验设备的控制各制造商应保证全部检验,测量和实验设备,包括机械,自动化或电子的检查和试验设备,适合于期望的目的,并有能力生产有价值的产物.各制造商应建立并保持关于保证常规校准,检验,检查和保养设备的程序.该程序应包括操作,防护和存储设备的的规定,以保持实用的精密度和准确性.有关内容均应记录在文件中.(b)校准校准程序应包括对准确度和精密度的准确说明和限值.当未达到准确度和精密度的限值时,应采取有效补救措施重建限值,并要评价是否对器械质量产生不利影响,有关内容要记录在文件中.(1)校准标准用于检验,测量和实验设备的校准标准应参照国家或国际标准.如果国家或国际标准不适用或不可得,制造商应使用一份自主的复制标准.如果没有可用的标准存在,制造商应建立并保持一份内部执行标准.(2)校准记录设备鉴定,校准日期,每次校准的执行人及下一次校准的日期,均应记录在文件中.这些记录应放在每台设备上(或附近),或者使用设备和校准设备的人都备有记录.§820.75 过程确认(a)当过程的结果不能被随后的检验和试验完全验证时,应建立高标准的保证和认可程序使加工过程确认.过程确认和结果,执行日期和执行人的签名,必要的设备,均应记录在文件中.(b)各制造商应建立并保持关于检测和控制确认过程的过程参数的程序,以保证持续达到指定的要求.(1)各制造商应保证由限定的人完成确认过程.(2)确认过程,监测和控制方法及数据,执行日期,必要时完成确认过程的操作者或使用的主要设备均应记录在文件中.(c)当过程确认发生变化或偏差时,制造商应检查并评价过程确认,必要时要使其再确认.有关内容应记录在文件中.1.8 认可行为§820.80 进货,加工过程和成品的认可(a)总则各制造商应建立并保持认可的程序.认可包括检验,试验或其他验证行为.(b)进货认可行为各制造商应建立并保持认可接受进厂产品的程序.对接受进厂的产品应进行检验,试验或其他验证以达到指定要求.认可和拒绝均应记录在文件中.(c)加工过程中产品的认可行为适当的时候,各制造商应建立并保持保证加工过程中的产品达到指定要求的认可程序.这种程序在完成要求的检验,试验或其他验证行为,或者收到必须的认可证明之前,应保证加工过程中产品控制,并记录在文件中.(d)成品认可行为各制造商应建立并保持认可成品的程序,以保证单个或各批成品达到认可标准.成品在认可以前应隔离放置,或以其他方式适当控制.成品在达到以下要求时,才可进行分发:完成DMR的要求;查阅相关数据和文件;指定专人批准许可并签名;注明批准日期.(e)认可记录各制造商应将认可行为记录在文件中.这些记录应包括:执行的认可行为,执行日期,结果,执行认可行为的个人签名,使用的适当设备.这些记录应作为DHR 的一部分内容.§820.86 认可状况各制造商应以适当的方式检验产品的认可状况,以指明产品是否符合认可标准.认可状况的检验应贯穿整个产品制造,包装,标签,安装和服务的过程,以保证只有通过认可的产品才能分发,使用或安装.1.9 不合格品§820.90 不合格品(a)不合格品控制各制造商应建立并保持控制不合格产品的程序.程序中应写明不合格品的标识,记录,评价,隔离和处置.不合格评价包括确定是否需要调查并告知责任人或机构.评价和调查均应记录在文件中.(b)不合格品的评审和处置(1)各制造商应建立并保持评审和批准处置不合格品的职责的程序.程序应阐明评审和处置过程.对不合格品的处置过程应记录在文件中.文件还包括某不合格品是可用的依据及批准人签名.(2)各制造商应建立并保持返工的程序,包括对不合格品返工之后的复试和复评,以保证产品达到现行的认可规范.返工和复评行为,包括确定返工对产品的不良影响,均应记录在DHR文件中.1.10 纠正和预防措施§820.100 纠正和预防措施各制造商应建立和保持实施纠正和预防措施的程序,程序应包括下列要求:(1)分析过程,操作,让步,质量审核报告,质量记录,服务记录,意见,返工产品或其他来源的数据,以查明导致不合格品或其他质量问题的现存和潜在原因.必要的时候,要适当使用统计学方法分析会再发生的质量问题.(2)调查与生产过程和质量体系有关的不合格原因.(3)确定纠正和防止再发生不合格品和其他质量问题的必须措施.(4)验证纠正和防止措施是否有效,并对成品器械无不利影响.(5)执行和记录修改的方法和程序,必须纠正和预防查明的质量问题.(6)保证与质量问题或不合格品有关的信息能传达给那些直接负责保证该产品质量或预防此类问题的有关人员.(7)把查明的质量问题的相关信息和纠正及预防措施提交管理机构评审.(8)纠正和预防措施的全部措施及结果均记录在文件中.1.11 标签和包装的控制§820.120 器械标签各制造商应建立和保持控制标签的程序.(a)标签完整标签的印刷和应用应保持完整,并且在加工,贮存,搬运,分发和使用过程中的物品均应有标签.(b)标签审查指定专人审查标签的准确性,若适用应包括正确的有效期,控制编号,储存说明,搬运说明和其他附加的处理说明.(c)标签存储各制造商应以能够正确鉴别标签的方式储存标签,并防止混乱.(d)标签操作各制造商应控制标签和包装操作以防止混乱.标签和标签操作的单个或批量产品均应记录在DHR文件中.(e)控制编号按§820.65中要求,控制编号应在整个分发过程中附在器械上.§820.130 器械包装各制造商应保证器械的包装和运输容器经一定设计和管理,能保护器械在加工,储存,搬运和分发的通常情况下不致改变或损坏.1.12 搬运,储存,分发和安装§820.140 搬运各制造商应建立并保持在搬运过程中防止发生混乱,损坏,变质,污染或其他对产品的不良影响的程序.§820.150 储存(a)各制造商应建立并保持控制产品储存场地和库房的程序,以防止混淆,损坏,变质,污染或其他在使用和分发以前的不利影响,并保证不使用或分发过期的,废弃的或变质的产品.为防止产品超过保质期,应以能促进货品轮流发送的方式储存,且货架条件应合适.(b)各制造商应建立并保持储存场地的授权接收或发送方法的程序.§820.160 分发(a)各制造商应建立并保持控制分发成品的程序,以保证只分发被认可的器械,不分发已到期或变质的器械,并检查订单,以保证在器械分发之前无混乱和错误.(b)各制造商应建立并保持分发记录,包括下述内容:最初运送的地址和姓名,装运器械的标识和数量,装运日期,器械的控制编号.§820.170 安装(a)需要安装器械的各制造商应建立并保持适当的安装和检查说明书及适当的试验程序.安装说明书和程序随器械一起分发或送给安装者,说明书和程序应包括正确安装的指导方法,使安装后器械能正常使用.。
美国FDA_医疗器械体系法规QSR820中英文版

美国FDA 医疗器械体系法规QSR820中文版Part 820——质量体系法规——目录Subpart A- 总则820.1 范围820.3 定义820.5 质量体系Subpart B –质量体系要求820.20 管理职责820.22 质量审核820.25 人员Subpart C- 设计控制820.30 设计控制Subpart D- 文件控制820.40 文件控制Subpart E- 采购控制820.50 采购控制Subpart F- 标识与可追溯性820.60 标识820.65 可追溯性Subpart G - 生产和过程控制820.70 生产和过程控制820.72 检验、测量和试验设备820.75 过程确认Subpart H - 验收活动:820.80 进货、过程和成品器械检验820.86 检验状态Subpart I –不合格品820.90 不合格品Subpart J - 纠正和预防措施820.100 纠正和预防措施Subpart K –标识和包装控制820.120 设备标签820.130 设备包装Subpart L –搬运/储存/分销和安装820.140 搬运820.150 贮存820.160 分销820.170 安装Subpart L –记录820.180 记录的通用要求820.181 设备主要记录820.184 设备历史记录820.186 质量体系记录820.198 投诉文件Subpart M –服务820.200 服务Subpart N –统计技术820.250 统计技术Subpart A——总则Subpart A--General ProvisionsSec.820.1 范围Sec. 820.1 Scope.(a)适用性Applicability。
(1)本质量体系法规阐明了当前良好制造法规Current good manufacturing practice (CGMP)的要求。
本标准适用于所有预期用于人类的成品器械的设计、制造、包装、标识、储存、安装和服务中所使用的管理方法、设施和控制。
美国FDA-医疗器械体系法规QSR820中英文版2015.06

培训教材美国FDA医疗器械体系法规QSR820中文版Part820820.1820.3820.5SubpartD-文件控制820.40 文件控制SubpartE-采购控制820.50 采购控制SubpartF-标识与可追溯性820.60 标识820.65 可追溯性SubpartG-生产和过程控制820.70 生产和过程控制820.72 检验、测量和试验设备 820.75 过程确认SubpartH-验收活动:SubpartI820.170 安装SubpartL–记录820.180 记录的通用要求820.181 设备主要记录820.184 设备历史记录820.186 质量体系记录820.198 投诉文件SubpartM–服务820.200 服务SubpartN–统计技术820.250 统计技术(a(1和控制品法案(2,(3(b规,包括本章此部分和其它部分的情况,特别是对讨论中的设备,此法规应取代其它通用要求。
伪劣产品。
这类产品及对此不符合负责的任何个人,将依法被起诉。
(d)外国制造商。
如果把器械进口到美国的制造商拒绝允许或同意FDA对其外国工厂履行为确定器械是否符合本法规(Part820)所进行的检查,可按section801(a)条款对其提出诉讼。
即准备出口到美国的设备,其设计、生产、包装、标签、贮存或服务中使用的方法和设备控制不符合本法令section520(f)和本部分(Part820)的要求,可按本法令section501(h)条款判定在此条件下制造的产品为伪劣产品。
(e)豁免或特别许可/Exemptionsorvariances(1)任何人希望得到任何(2)在有关部门确定此种改变符合美国公众健康的最佳利益时,FDA可能发起并同意器械质量体系的特别许可。
公在美国公众健康确实需要该设备,且如无此特别许可,则器械就不可能充分有效的生产的情况下,特别许可才有效。
(2)FDAmayinitiateandgrantavariancefromanydevicequalitysystemrequirementwhentheagencydeterminesthatsuchva rianceisinthebestinterestofthepublichealth.Suchvariancewillremainineffectonlysolongasthereremainsapublichealthne edforthedeviceandthedevicewouldnotlikelybemadesufficientlyavailablewithoutthevariance.(f)本部分不适用于本章897部分定义的烟草销售商。
FDA质量体系规范中文版(QSR820).

FDA 质量体系规范中文版 (QSR8201.1 概述§820.1 范围(a 适用性(1 在这个质量体系规范中描述了现行的生产管理规范的要求 (CGMP. 本规范要求规定了所有医用器械成品在设计 , 制造 , 包装 , 标签 , 贮存 , 安装和服务中使用的方法 , 设施和控制 . 这些要求是为了确保医疗器械成品的安全和有效 , 并遵从美国食品 , 药品和化妆品法 . 本规范提出了适用于医疗器械成品制造商的基本要求 . 如果某制造商只进行本规范规定的一部分操作 , 而不进行其他操作 , 则该制造商仅需执行适用于他所进行操作的那些要求 . 有关Ⅰ类器械 , 设计控制仅按在§820.30(a(2中列出的要求进行 . 这个规范不适用于成品组件和零件的制造商 , 但鼓励这样的制造商使用规范的适当规定作为指导 . 人类血液制品和血液成分的制造商不属于本规范的管理范围 , 但属于 606的管理范围 .(2 这一规范的规定适用于本规范定义的医疗器械成品 , 即使用的对象是人的 , 在美国各州或领地 , 哥伦比亚特别区或波多黎哥联邦制造 , 进口或出口的器械成品 .(3 在本规范中 , 几次使用了短语”适当的地方”. 当要求以”适当的地方”来限制时 , 如果制造商没有合理的理由来证明不适宜 , 就认为此要求是”适当的”. 如果不贯彻”适当的”要求 , 就会导致产品达不到要求或制造商不能采取某些必要的正确措施 .(b 范围这一的质量体系规范和增补在这一规范其他节的条文明确注明用于其他方面的条文除外 . 在不可能执行全部适用条文 (包括这一节的和这一规范其他节的条文的情况下 , 特定的运用于有问题器械的条文将取代其他一般的适用条文 .(c 权威性根据联邦法 501, 502, 510, 513, 514, 515, 518, 519, 520, 522, 701, 704, 801, 803, (21U.S.C.315, 352, 360, 360c, 360d, 360e, 360h, 360i, 360j, 360l, 371, 374,381, 383 建立和提出了 820规范的权威性 . 如果没能执行适用规范 , 可能会导致产生伪劣器械 , 根据法规 501(h, 对这样的器械和未能执行规范的人都要进行处罚 .(d 外国制造商如果某制造商提供给美国的进口器械 , 拒绝接受 FDA 对外国设备进行是否执行本规范的检查 , 就会出现法规的 801(a节中的后果 . 用这样的设备生产出的任何器械 , 在设计 , 制造 , 包装 , 标签 , 贮存 , 安装或服务方面 , 使用方法 , 设备和控制都未遵从法规的 520(f节和本规范的要求 . 根据法规 501(h, 用这样的设备制造出来的器械都属于伪劣产品 . (e 豁免或更改(1 任何希望豁免或更改执行某些器械质量体系要求的申请 , 都要遵从法规520(f(2的要求 . 申请豁免或更改的过程将依据这一规范§10.30的程序进行 , 即 FDA 的管理程序 . 以下地址可提供指导 : 器械和放射卫生中心 , 小制造商处 (HFZ-220, 1350 Picca rd Dr., Rockville, MD20850, U.S.A. 电话 : 1-800-638-2041或 1-301-443-6597, FAX 301-443-8818. (2 当 FDA 判定某种更改有益于公众健康时 , 就会起草并认可这项更改 . 这种更改仅能在一段时间内维持有效 , 即当器械仍能满足公众健康需要 , 并且如果没有更改 , 器械不可能制造得非常有效的一段时间内 .§820.3 定义(a法指的是美国食品 , 药品和化妆品法修正案 (secs.201~903, 52Stat.1040 et seq., 修正版 21U.S.C.321~394. 在法 201中的全部定义都适用于本规定 .(b投诉指的是以某些书面的 , 电子的或口头的形式表达意见 , 认为在配发后的器械在鉴定 , 质量 , 耐久性 , 可靠性 , 安全性 , 有效性或性能方面有缺陷 .(c组成指的是原材料 , 物质 , 小件 , 零件 , 软件 , 硬件 , 标签或有包装和标签的成品器械的零配件 .(d控制编号指的是有区别的符号 , 如字母或数字的不同组合 , 或以原制造 , 包装 , 标签和分发的单个或批量成品的区别符号来分辨 .(e设计历史文件 (DHF指的是描述某医疗器械成品设计过程的有关记录 .(f设计输入是指作为器械设计基础对器械的物理和特性要求 .(g设计输出是指各设计阶段的设计成果和最终的总设计成果 . 完成设计输出包括器械 , 包装和标签 , 器械主记录 .(h设计评审是指依照依照文件进行广泛 , 系统的设计评审 , 以评价设计要求的适当性 , 并评价设计达到这些要求的能力 , 查明问题所在 .(i器械历史记录 (DHR是指医疗器械成品制造过程的记录 .(j器械主记录 (DMR是指包括医疗器械成品的程序和规范的完满记录 .(k建立是指定义 , 文件 (书面的或电子的和执行情况 .(l器械成品是指适于使用或具有功能的器械或器械附件 , 不论是否经过包装 , 贴标签或灭菌 .(m批是指一种或几种组成或成品器械具有单一类型 , 型号 , 类别 , 尺寸 , 成分或软件版本 , 必须在相同条件下制造 , 并在规定的限度内具有相同的特征和质量 .(n管理职责是指制造商的高级雇员有权建立或改变制造商的质量方针和质量体系 .(o制造商是指设计 , 制造 , 构造 , 装配或加工成品器械的人 . 制造商包括但不局限于那些从事灭菌 , 安装 , 再贴标签 , 再制造 , 再包装或Specification 开发商和从事这些工作的外国实体的最初代理人 .(p生产过程副产物是指促进生产过程所用的材料或物质 , 制造加工过程中的伴随组分或副产品 , 以残余物或混杂物的形式存在 .(q不合格是指未达到特定的要求 .(r产品是指组成 , 制造材料 , 加工过程中器械 , 成品器械及返回器械 . (s质量是指使器械安全适用的总性质和特征 , 包括安全性和性能 . (t质量审核是指在规定的时间间隔 , 以足够的次数 , 对制造商质量体系进行有组织的自主的检查 , 检验质量体系行为和结果是否执行质量体系程序 , 以保证有效地执行程序 , 达到质量体系目标 .(u质量方针是指有关质量的机构方向和目标 , 是由负责的管理人员建立的 .(v质量体系是指检查质量管理的组织机构 , 职责 , 程序 , 处理和资源 . (w再加工是指对成品器械进行加工 , 调节 , 革新 , 再包装 , 再贮存 , 大大改变了成品器械的性能 , 安全性规范或用途 .(x返工指的是对不合格产品采取某些措施 , 以使他在获准配发之前达到指定的 DMR 要求 .(y规范是指生产 , 加工 , 服务或其他行为必须遵守的一些要求 . (z有效性是指通过检查和提供客观证据来证明能始终满足特定的用途 .(1过程确认是指通过客观的证据证明加工生产出的产物或产品始终达到预定的规范 .(2设计确认是指通过客观的证据证明器械规范与使用者的需要和设计的用途相一致 .(aa验证是指通过检查和提供客观证据来证明已经满足指定的要求 . §820.5 质量体系各制造商应建立并保持一个质量体系 , 适合于他们设计或制造的医疗器械 , 并且达到本规范的要求 .1.2 质量体系要求§820.20 管理职责(a质量方针管理职能机构应建立质量方针目标和质量承诺 , 并保证质量方针在企业各级人员中的理解 , 贯彻和持续执行 .(b管理机构各制造商都应建立并维持一个适当的组织机构 , 以保证器械依照本规范进行设计和生产 .(1职责和权限各制造商都应任命有相应职责 , 权限和能独立行使职权的人员负责管理 , 执行和评价质量体系 .(2人员各制造商都应具备足够的合格人员 , 包括分派培训有素的人员从事管理 , 执行 , 评价和内部质量审核等工作 , 以达到本规范要求 . (3管理者代表管理职能机构应任命其中一员为管理者代表 , 并在文件中注明 . 管理者代表不论其他职责如何 , 必须履行下列职责和权力 :i. 确保按本规范要求有效地建立和保持 .ii. 向管理机构汇报质量体系进行情况 , 供其讨论 .(c管理评审管理职能机构应按照建立的程序 , 以足够的次数定期评审质量体系的适用性和有效性 . 以保证质量体系达到本规范要求和制造商建立的质量方针和目标 , 评审日期和结果应形成文件 .(d质量策划各制造商应编制质量计划 , 确定与设计和制造的器械相关的质量实践 , 人员和措施 , 并建立达到质量要求的规划 .(e质量体系程序各制造商应建立质量体系的各种程序和实施指南 , 并形成文件 .§820.22 质量审核各制造商应建立质量审核的程序 , 并进行管理 , 以保证质量体系符合建立的质量体系要求 , 确定该质量体系的有效性 . 质量审核应由与审核事物无直接责任的人执行 . 若有必要时 , 应采取措施纠正错误措施 , 包括对有缺陷的事物进行再审核 .管理机构对各质量审核的结果及再审核的情况进行复核 . 提供质量审核日期和结果及再审核的有关文件 .§820.25 全体工作人员(a一般要求各制造商都应具有足够的工作人员 , 具备必需教育 , 背景 , 接受过培训并富有经验 , 以保证正确履行本节所要求的全部工作 .(b培训各制造商应建立必需培训的程序 , 保证全部工作人员在经过培训后能胜任他们各自的职责 , 并提供与培训有关的文件 .(1培训内容还包括使全体工作人员懂得由于错误执行指定工作可能会导致器械产生缺陷 .(2使从事验证和确认工作的全体工作人员能预见可能会发生的缺陷和错误 .1.3 设计控制§820.30 设计控制(a 总则(1 Ⅱ,III 类器械的制造商 , 以及在本规范 (a(2段列出的Ⅰ类器械制造商 , 应建立并保持控制器械设计的方法 , 以保证达到特定的要求 .(2 下列Ⅰ类器械也需要设计控制 .i. 计算机软件的自动化机械 .ii. 下面列出的器械 :(b设计和开发计划各制造商应建立并保持有关设计和开发行为的计划 , 并规定执行职责 . 计划应规定提供或输入设计和开发程序的不同组或行为的互换信息 . 对计划应进行检查 , 用现代化手段处理并证实设计和开发的进展 .(c设计输入各制造商应建立并保持关于保证器械的设计要求适当的程序 , 以器械用途为主 , 包括使用者和病人的需要 . 该程序应包括关于不完善 , 不清楚或抵触要求的处理办法 . 设计输入要求应记录在文件中 , 并由指定的人进行检查和认可 , 并提供认可这些要求的日期和个人签名的文件 . (d设计输出各制造商应建立并保持关于确定和提供设计输出文件的程序 , 并进行执行设计输入要求的适当评价 . 设计输出程序应包含或制定参照的认可标准 , 并保证那些设计输出是鉴定器械良好性能所必需的 . 设计输出应记录在文件中 , 在获准之前进行评审 , 并提供有关评审认可日期和签名的文件 .(e设计评审各制造商应建立并保持一套程序 , 保证在器械设计开发的适当阶段 , 按计划评审设计结果 , 并提供正式文件 . 评审参加者应包括设计的专业人员对设计阶段负有责任的代表和与设计阶段五直接责任的人和必要的专家 . 设计评审的结果包括设计鉴定 , 评审人员和日期 , 都应记录在设计历史文件 (DHF中 .(f设计验证各制造商应建立并保持验证器械设计的程序 . 设计验证应证明设计输出达到设计输入要求 . 设计验证的结果 , 包括设计方法的鉴定 , 验证人员和日期 , 都应当记录在 DHF 文件中 .(g设计确认各制造商应建立并保持设计确认的程序 . 应在规定的操作条件下 , 对试制的单个 , 批量产品或等同物进行设计确认的确认 . 设计确认应保证器械满足使用者的需要 , 并具有预期用途 , 还应包括产品在实际或设想使用条件下的试验 . 设计确认还应包括软件确认及适当的时候的风险分析 . 有关设计确认的结果 , 包括对设计和设计方法的鉴定 , 执行人员和日期都应记录在 DHF 文件中 .(h设计转换各制造商应建立并保持一套程序以确保器械设计正确性体现在一定的生产规范中 .(i设计更改各制造商应建立并保持一套程序 , 对更改的设计在执行之前进行鉴定 , 提供有效性文件或适当的地方进行验证 , 评审和认可 . (j设计历史文件各制造商应建立并保持各种类型器械的 DHF. DHF应包含或参照必要的原始记录 , 来证明设计开发过程与认可的设计计划一致 , 并遵守本规范要求 .1.4 文件控制§820.40 文件控制各制造商应建立并保持本规范所要求的全部文件控制的程序 . 程序应提供下列内容 :(a文件认可和发布各制造商应在分发达到本规范要求的全部文件之前 , 委派专人检查适用性和认可情况 . 应提供有关认可文件的日期和个人签名的文件 . 达到本规范要求的文件适用于指定的 , 使用的或其他需要的地方 , 所有失效的文件应从使用条款中删除 .(b文件更改更改文件应由执行原文件检查和认可的同一职能部门内的人进行检查和认可 , 除非有另外明确指定人选 . 认可的改动应及时地转达给有关人员 . 各制造商应保留更改文件的记录 . 更改记录应包括修改内容 , 相关文件的鉴定 , 认可人的签名 , 认可日期及更改生效的日期 .1.5 采购控制§820.50 采购控制各制造商应建立并保持确保所有购买的或收到的产品和服务符合指定要求的程序 .(a对供应商 , 承包商和咨询机构的评审各制造商应建立一套供应商 , 承包商和咨询机构必须达到的指定要求 . 各制造商应 :(1根据指定要求 (包括质量要求 , 评价和选择潜在的供应商 , 承包商和咨询机构 . 评价应记录在文件中 .(2根据评价结果 , 确定对产品 , 服务 , 供应商 , 承包商和咨询机构实施控制的方式和程度 .(3建立和保持可接受的供应商 , 承包商和咨询机构的记录 .(b采购资料各制造商应对采购的或收到的产品和服务建立并保留关于是否达到质量要求的资料 . 可能的话 , 应包括一份协议 , 关于供应商 , 承包商和咨询机构同意告知制造商 , 他们的产品或服务的改变 , 是制造商可以判断这些改变是否会影响成品器械的质量 . 采购资料应依照§820.40得到认可 .1.6 标识和可追溯性§820.60 标识各制造商为防止混乱应建立并保持在接收 , 制造 , 交付和安装各阶段的产品标识程序 .§820.65 可追溯性对于生产外科植入人体 , 支持或维持生命的器械制造商和依照制造商提供的使用说明正确使用时 , 如果器械运行失败可对使用者造成严重伤害 , 则应建立并保持对每个或每批产品都有唯一性标识的程序 . 程序应促进纠正错误措施 . 这种标识应包括在设计历史文件中 .1.7 生产和过程控制§820.70 生产和过程控制(a总则各制造商应制定 , 实施 , 控制并监测生产过程 , 以保证器械遵守本规范 . 在制造加工过程中可能会发生违反规范的地方 , 制造商应建立并保持必须的生产过程控制的程序 , 生产过程控制应包括 :(1提供指导文件 , 标准操作程序(SOP’s,限定方法和生产控制方式 ;(2在生产过程中监测和控制加工参数和产品特征 ;(3应遵守的指定参考标准或编号 ;(4加工和加工设备的认可 ;(5工艺要求应阐述在工艺文件中或用通过鉴定和认可的代表性样品来表现 .(b生产和过程的改变各制造商应建立并保持改变规则 , 方法 , 加工或步骤的程序 . 这些改变在执行之前应被验证或在适当时依照§820.75使改变有效 , 这些行为均应记录在文件中 . 改变应依照§820.40得到认可 . (c环境控制在有理由认为周围环境条件对产品质量有不利影响时 , 制造商应建立并保持适当控制环境条件的程序 . 应定期检查环境控制系统 , 以核实该系统 , 包括必需设备的适当性 , 并正发挥着良好作用 . 检查应记录在文件中 .(d工作人员如果有理由认为工作人员和产品或环境的接触对产品质量有不利影响时 , 各制造商应建立并保持对工作人员的健康 , 卫生习惯 , 行为和衣着的要求 . 各制造商应保证在指定的环境下临时工作的其他人员接受适当的训练或由接受过训练的人进行监督 .(e污染控制各制造商应建立并保持防止对产品质量有不良影响的物质污染设备或产品的程序 .(f厂房应该设计适当厂房 , 具有足够的空间进行必须的操作 , 以防止混乱 , 并保证有序的操作 .(g设备各制造商应保证在制造加工过程中使用的全部设备都达到指定要求 ,并经过适当设计 , 建造 , 放置和安装以利于保养 , 调试 , 清洁和使用 . (1保养计划表各制造商应建立并保持调试 , 清洁和其他设备保养的计划表 , 以保证达到生产规范 . 保养行为 , 包括执行保养行为的日期和人员应记录在文件中 .(2检查各制造商应依照建立的程序进行定期检查 , 以保证完成设备保养计划 . 检查日期和执行人员应记录在文件中 .(3调试各制造商应将设备调整限度和允许公差的说明放在需要定期调试的设备商 (或附近 , 或者从事这些调试的工作人员都备有说明 .(h加工过程的副产物在有理由认为某加工过程的副产物对产品质量具有不利影响的情况下 , 各制造商应建立并保持使用和排除这种副产物的程序 , 以保证他被排除或减少到不会对产品质量有不利影响的量 . 排除或减少加工过程的副产物均应记录在在文件中 .(i自动化处理对于生产或质量体系所用的计算机或自动化数据处理系统 , 制造商应依照已签订的协议书验证计算机软件是否具有预想的用途 . 修改的软件验证有效后方能批准和发布 . 验证过程和结果应记录在文件中 . §820.72 检验 , 测量和实验设备(a检验 , 测量和实验设备的控制各制造商应保证全部检验 , 测量和实验设备 , 包括机械 , 自动化或电子的检查和试验设备 , 适合于期望的目的 , 并有能力生产有价值的产物 . 各制造商应建立并保持关于保证常规校准 , 检验 , 检查和保养设备的程序 . 该程序应包括操作 , 防护和存储设备的的规定 , 以保持实用的精密度和准确性 . 有关内容均应记录在文件中 .(b校准校准程序应包括对准确度和精密度的准确说明和限值 . 当未达到准确度和精密度的限值时 , 应采取有效补救措施重建限值 , 并要评价是否对器械质量产生不利影响 , 有关内容要记录在文件中 .(1校准标准用于检验 , 测量和实验设备的校准标准应参照国家或国际标准 . 如果国家或国际标准不适用或不可得 , 制造商应使用一份自主的复制标准 . 如果没有可用的标准存在 , 制造商应建立并保持一份内部执行标准 .(2校准记录设备鉴定 , 校准日期 , 每次校准的执行人及下一次校准的日期 , 均应记录在文件中 . 这些记录应放在每台设备上 (或附近 , 或者使用设备和校准设备的人都备有记录 .§820.75 过程确认(a当过程的结果不能被随后的检验和试验完全验证时 , 应建立高标准的保证和认可程序使加工过程确认 . 过程确认和结果 , 执行日期和执行人的签名 , 必要的设备 , 均应记录在文件中 .(b各制造商应建立并保持关于检测和控制确认过程的过程参数的程序 , 以保证持续达到指定的要求 .(1各制造商应保证由限定的人完成确认过程 .(2确认过程 , 监测和控制方法及数据 , 执行日期 , 必要时完成确认过程的操作者或使用的主要设备均应记录在文件中 .(c当过程确认发生变化或偏差时 , 制造商应检查并评价过程确认 , 必要时要使其再确认 . 有关内容应记录在文件中 .1.8 认可行为§820.80 进货 , 加工过程和成品的认可(a总则各制造商应建立并保持认可的程序 . 认可包括检验 , 试验或其他验证行为 .(b进货认可行为各制造商应建立并保持认可接受进厂产品的程序 . 对接受进厂的产品应进行检验 , 试验或其他验证以达到指定要求 . 认可和拒绝均应记录在文件中 .(c加工过程中产品的认可行为适当的时候 , 各制造商应建立并保持保证加工过程中的产品达到指定要求的认可程序 . 这种程序在完成要求的检验 , 试验或其他验证行为 , 或者收到必须的认可证明之前 , 应保证加工过程中产品控制 , 并记录在文件中 .(d成品认可行为各制造商应建立并保持认可成品的程序 , 以保证单个或各批成品达到认可标准 . 成品在认可以前应隔离放置 , 或以其他方式适当控制 . 成品在达到以下要求时 , 才可进行分发 :完成 DMR 的要求 ; 查阅相关数据和文件 ; 指定专人批准许可并签名 ; 注明批准日期 .(e认可记录各制造商应将认可行为记录在文件中 . 这些记录应包括 :执行的认可行为 , 执行日期 , 结果 , 执行认可行为的个人签名 , 使用的适当设备 . 这些记录应作为 DHR 的一部分内容 .§820.86 认可状况各制造商应以适当的方式检验产品的认可状况 , 以指明产品是否符合认可标准 . 认可状况的检验应贯穿整个产品制造 , 包装 , 标签 , 安装和服务的过程 , 以保证只有通过认可的产品才能分发 , 使用或安装 .1.9 不合格品§820.90 不合格品(a不合格品控制各制造商应建立并保持控制不合格产品的程序 . 程序中应写明不合格品的标识 , 记录 , 评价 , 隔离和处置 . 不合格评价包括确定是否需要调查并告知责任人或机构 . 评价和调查均应记录在文件中 . (b不合格品的评审和处置(1各制造商应建立并保持评审和批准处置不合格品的职责的程序 . 程序应阐明评审和处置过程 . 对不合格品的处置过程应记录在文件中 . 文件还包括某不合格品是可用的依据及批准人签名 .(2各制造商应建立并保持返工的程序 , 包括对不合格品返工之后的复试和复评 , 以保证产品达到现行的认可规范 . 返工和复评行为 , 包括确定返工对产品的不良影响 , 均应记录在 DHR 文件中 .1.10 纠正和预防措施§820.100 纠正和预防措施各制造商应建立和保持实施纠正和预防措施的程序 , 程序应包括下列要求 :(1分析过程 , 操作 , 让步 , 质量审核报告 , 质量记录 , 服务记录 , 意见 , 返工产品或其他来源的数据 , 以查明导致不合格品或其他质量问题的现存和潜在原因 . 必要的时候 , 要适当使用统计学方法分析会再发生的质量问题 .(2调查与生产过程和质量体系有关的不合格原因 .(3确定纠正和防止再发生不合格品和其他质量问题的必须措施 .(4验证纠正和防止措施是否有效 , 并对成品器械无不利影响 .(5执行和记录修改的方法和程序 , 必须纠正和预防查明的质量问题 .(6保证与质量问题或不合格品有关的信息能传达给那些直接负责保证该产品质量或预防此类问题的有关人员 .(7把查明的质量问题的相关信息和纠正及预防措施提交管理机构评审 .(8纠正和预防措施的全部措施及结果均记录在文件中 .1.11 标签和包装的控制§820.120 器械标签各制造商应建立和保持控制标签的程序 .(a标签完整标签的印刷和应用应保持完整 , 并且在加工 , 贮存 , 搬运 , 分发和使用过程中的物品均应有标签 .(b标签审查指定专人审查标签的准确性 , 若适用应包括正确的有效期 , 控制编号 , 储存说明 , 搬运说明和其他附加的处理说明 .(c标签存储各制造商应以能够正确鉴别标签的方式储存标签 , 并防止混乱 .。
美国FDA_医疗器械体系法规QSR820中英文版

美国FDA 医疗器械体系法规QSR820中文版之南宫帮珍创作Part 820——质量体系法规——目录Subpart A- 总则820.1 范围820.3 界说 820.5 质量体系Subpart B –质量体系要求820.20 管理职责820.22 质量审核 820.25 人员Subpart C- 设计控制 820.30 设计控制Subpart D- 文件控制 820.40 文件控制Subpart E- 推销控制 820.50 推销控制Subpart F- 标识与可追溯性820.60 标识 820.65 可追溯性Subpart G - 生产和过程控制820.70 生产和过程控制820.72 检验、丈量和试验设备 820.75 过程确认Subpart H - 验收活动: 820.80 进货、过程和制品器械检验 820.86 检验状态Subpart I –分歧格品 820.90 分歧格品Subpart J - 纠正和预防办法 820.100 纠正和预防办法Subpart K –标识和包装控制820.120 设备标签 820.130 设备包装Subpart L –搬运/贮存/分销和装置820.140 搬运820.150 贮存820.160 分销 820.170 装置Subpart L –记录820.180 记录的通用要求820.181 设备主要记录820.184 设备历史记录820.186 质量体系记录 820.198 投诉文件Subpart M –服务 820.200 服务Subpart N –统计技术820.250 统计技术Subpart A——总则Subpart A--General Provisions Sec.820.1 范围Sec. 820.1 Scope. (a)适用性Applicability. (1)实质量体系法规说明了以后良好制造法规Current good manufacturing practice(CGMP)的要求.本标准适用于所有预期用于人类的制品器械的设计、制造、包装、标识、贮存、装置和服务中所使用的管理方法、设施和控制.本标准的目的是保证制品器械的平安性和有效性, 并符合联邦食品、药品和化妆品法案Federal Food, Drag and Cosmetic Act (the act).本法规适用于所有的医疗器械制品制造商.如果制造商仅从事本部份有要求服从的某些过程而未从事其它过程, 则只需符合其实施的过程的要求.对Ⅰ类设备, 设计控制仅适用于Sec.820.30(a)(2)中列出的设备.本法规不适用于制品器械的部件或零件制造商, 但鼓励这类制造商把本法规的适当规定作为指南来使用.人血和血液成份制造商不受本部份法规的限制, 但应遵循本章606部份法规的要求.Current good manufacturing practice(CGMP) requirements are set forth in this quality system regulation. The requirements in this part govern the methods used in, and the facilities and controls used for, the design, manufacture, packaging, labeling, storage, installation, and servicing of all finished devices intended for human use. The requirements in this part are intended to ensurethat finished devices will be safe and effective and otherwise in compliance with the Federal Food, Drug, and Cosmetic Act (the act). This part establishes basic requirements applicable to manufacturers of finished medical devices. If a manufacturer engages in only some operations subject to the requirements in this part, and not in others, that manufacturer need only comply with those requirements applicable to the operations in which it is engaged. With respect to class I devices, design controls apply only to those devices listed in 820.30(a)This regulation does not apply to manufacturers of components or parts of finished devices, but such manufacturers are encouraged to use appropriate provisions of this regulation as guidance. Manufacturers of human blood and blood components are not subject to this part, but are subject to part 606 of this chapter. Manufacturers of human cells, tissues, and cellular and tissue-based products (HCT/Ps), as defined in 1271..3(d) of this chapter, that are medical devices (subject to premarket review or notification, or exempt from notification, under an application submitted under the device provisions of the act or under a biologicalproduct license application under section 351 of the Public Health Service Act) are subject to this part and are also subject to the donor-eligibility procedures set forth in part 1271 subpart C of this chapter and applicable current good tissue practice procedures in part 1271 subpart D of this chapter. In the event of a conflict between applicable regulations in part 1271 and in other parts of this chapter, the regulation specifically applicable to the device in question shall supersede the more general.(2)本部份的规定适用于本部份界说的预期用于人体的所有制品器械, 不论其在美国(包括:美国任何州或领土,哥伦比亚特区,波多黎各联邦)外乡制造还是进口,提供进口的产物.(2) The provisions of this part shall be applicable to any finished device as defined in this part, intended for human use, that is manufactured, imported, or offered for import in any State or Territory of the United States, the District of Columbia, or the Commonwealth of Puerto Rico.(3)在本法规中“适用时”(where appropriate)呈现过屡次.当要求根据“where appropriate”被认为是合格时, 其要求应被认为是“适用的”(appropriate), 除非组织能提供文件证明其理由.如果不执行预期结果会招致产物不符合其特定的要求, 或组织不需要执行任何需要的纠正办法, 那么要求就是适用的(appropriate).(3) In this regulation the term "where appropriate" is used several times. When a requirement is qualified by "where appropriate," it is deemed to be "appropriate" unless the manufacturer can document justification otherwise. A requirement is "appropriate" if non-implementation could reasonably be expected to result in the product not meeting its specified requirements or the manufacturer not being able to carry out any necessary corrective action.(b)限制.除非特别规定, 则本部份质量体系法规是本章其它部份法规的弥补要求.在不能符合所有适用的法规, 包括本章此部份和其它部份的情况, 特别是对讨论中的设备, 此法规应取代其它通用要求.(b) The quality system regulation in this part supplements regulations in other parts of this chapterexcept where explicitly stated otherwise. In the event of a conflict between applicable regulations in this part and in other parts of this chapter, the regulations specifically applicable to the device in question shall supersede any other generally applicable requirements.(c)权限.PART820是在(21U.S.C.法令351、352、360、360c、360d、360e、360h、360i、360j、360l、370、374、381、383中)501、502、510、513、514、515、518、519、520、522、701、704、801、803下建立并发布的.不符合本部份(Part 820)的任何适用的规定, 依据法令section 501(h)条款, 可判定该产物为伪劣产物.这类产物及对此不符合负责的任何个人, 将依法被起诉.(c)Authority.Part 820 is established and issued under authority of sections 501, 502, 510, 513, 514, 515, 518, 519, 520, 522, 701, 704, 801, 803 of the act (21 U.S.C. 351, 352, 360, 360c, 360d, 360e, 360h, 360i, 360j, 360l, 371, 374, 381, 383). The failure to comply with any applicable provision in this part renders a device adulterated under section 501(h) of the act. Such a device, as well as any person responsible for the failure to comply, is subject to regulatory action.(d)外国制造商.如果把器械进口到美国的制造商拒绝允许或同意FDA对其外国工厂履行为确定器械是否符合本法规(Part 820)所进行的检查, 可按section 801(a)条款对其提出诉讼.即准备出口到美国的设备, 其设计、生产、包装、标签、贮存或服务中使用的方法和设备控制不符合本法令section 520(f)和本部份(Part 820)的要求, 可按本法令section 501(h)条款判定在此条件下制造的产物为伪劣产物.(d)Foreign manufacturers.If a manufacturer who offers devices for import into the United States refuses to permit or allow the completion of a Food and Drug Administration (FDA) inspection of the foreign facility for the purpose of determining compliance with this part,it shall appear for purposes of section 801(a) of the act, that the methods used in, and the facilities and controls used for, the design, manufacture, packaging, labeling, storage, installation, or servicing of any devices produced at such facility that are offered for import into the United States do not conform to the requirements of section 520(f) of the act and this part and that the devices manufactured at that facility are adulterated under section 501(h) of the act.(e)豁免或特别许可/Exemptions or variances (1)任何人希望得就任何医疗器械质量体系要求的豁免或特别许可, 应符合法令section 520(f)(2)的要求.根据本章Sec.10.30即FDA行政法式, 来提交豁免或特别许可的申请.可以从器械和辐射健康中心和小型制造商援助处获得指导, 地址(HFZ-220), 1350 Piccard Dr., Rockville, MD20850, U.S.A., 德律风1-800-638-2041或1-301-443-6597, 传真301-443-8818. (1) Any person who wishes to petition for an exemption or variance from any device quality system requirement is subject to the requirements of section 520(f)(2) of the act. Petitions for an exemption or variance shall be submitted according to the procedures set forth in 10.30 of this chapter, the FDA's administrative procedures. Guidance is available from the Center for Devices and Radiological Health, Division of Small Manufacturers, International and Consumer Assistance (HFZ-220), 1350 Piccard Dr., Rockville, MD20850, U.S.A., telephone 1-800-638-2041 or 240-276-3150, FAX 240-276-3151.(2)在有关部份确定此种改变符合美国公众健康的最佳利益时, FDA可能发起并同意器械质量体系的特别许可.公在美国公众健康确实需要该设备, 且如无此特别许可, 则器械就不成能充沛有效的生产的情况下, 特别许可才有效.(2) FDA may initiate and grant a variance from any device quality system requirement when the agency determines that such variance is in the best interest of the public health. Such variance will remain in effect only so long as there remains a public health need for the device and the device would not likely be made sufficiently available without the variance.(f)本部份不适用于本章897部份界说的烟草销售商.Sec.820.3 界说/ Definitions (a)法案Act.指明Federal Food, Drug and Cosmetic Act, 如修正的(secs.201-903, 52 Stat. 1040 et sep., 21 U.S.C. 321-394).所有法案section 201中的界说在本部份法规中均适用.(a)Act means the Federal Food, Drug, and Cosmetic Act, as amended (secs. 201-903, 52 Stat. 1040et seq., as amended (21 U.S.C. 321-394)). All definitions in section 201 of the act shall apply to the regulations in this part.(b)投诉Complaint.在设备交付后所有的书面的、电子的或口头的, 对设备的标识、质量、耐用性、可靠性、平安性、有效性和性能方面缺陷的信息.(b)Complaint means any written, electronic, or oral communication that alleges deficiencies related to the identity, quality, durability, reliability, safety, effectiveness, or performance of a device after it is released for distribution.(c)部件Component.所有意图用来包括成为已完成的、包装、标识的器械的一部份的原资料、物资、构件、零件、软件、固件、连接件、标签或它们的集合.(c)Component means any raw material, substance, piece, part, software, firmware, labeling, or assembly which is intended to be included as part of the finished, packaged, and labeled device.(d)控制号Control number.任何鉴别性符号, 如由字母、数字或它们的组合形成的唯一性组合, 由控制号可以确定一批或一个器械的制造、包装、标识和交付的历史.(d)Control number means any distinctive symbols, such as a distinctive combination of letters or numbers, or both,from which the history of the manufacturing, packaging, labeling, and distribution of a unit, lot, or batch of finished devices can be determined.(e)设计历史文件Design history file(DHF).制品器械的设计历史记录的汇总.(e)Design history file(DHF) means a compilation of records which describes the design history of a finished device.(f)设计输入Design input.器械实体和性能要求, 是产物设计的基础.(f)Design input means the physical and performance requirements of a device that are used as a basis for device design.(g)设计输出Design output.是指每个设计阶段和最后所有的设计功效的结果.已完成的设计输出是器械主记录的基础.全部最终完成的设计输出, 由器械及其包装和标识和设备主记录组成. (g)Design output means the results of a design effort at each design phase and at the end of the total design effort. The finished design output is the basis for thedevice master record. The total finished design output consists of the device, its packaging and labeling, and the device master record.(h)设计评审Design review.是指对设计的一个文件化的、全面的、系统的检查, 评价其满足设计要求, 评价其有能力满足要求, 并识别任何问题.(h)Design review means a documented, comprehensive, systematic examination of a design to evaluate the adequacy of the design requirements, to evaluate the capability of the design to meet these requirements, and to identify problems.(i)设备历史记录Device history record(DHR).制品器械历史记录的汇总.(i)Device history record(DHR) means a compilation of records containing the production history of a finished device.(j) Device master record(DMR).制品器械的法式和规范的汇总.(j)Device master record(DMR) means a compilation ofrecords containing the procedures and specifications for a finished device.(k)建立Establish.界说文件(书面或电子的)并执行.(k)Establish means define, document (in writing or electronically), and implement.(l)制品器械Finished device.设备或其附件, 无论其是否包装、标识或灭菌, 能够满足使用要求或者说能够实现其功能.(l)Finished device means any device or accessory to any device that is suitable for use or capable of functioning, whether or not it is packaged, labeled, or sterilized.(m) Lot或batch.一个或多个元件或制品器械, 均为同一种规格、型号、尺寸、成份或软件版本, 在相同条件下生产, 满足相同的特性和质量要求.(m)Lot or batch means one or more components or finished devices that consist of a single type, model, class, size, composition, or software version that are manufactured under essentially the same conditions and that are intended to have uniform characteristics and quality within specified limits.(n)执行职责的管理者Management with executive responsibility.是组织的高级员工, 他们负有建立或更改组织的质量方针和质量体系的职权.(n)Management with executive responsibility means those senior employees of a manufacturer who have the authority to establish or make changes to the manufacturer's quality policy and quality system.(o)制造商/组织Manufacturer.是指设计、制造、制作(fabricate)、装配或加工制品器械的任何人.制造商包括但不单限于根据合同执行灭菌、装置、重新标识、重新制造、重新包装或特定的开发职责的制造商, 和执行这些职责的国外组织的国内分销商.(o)Manufacturer means any person who designs, manufactures, fabricates, assembles, or processes a finished device. Manufacturer includes but is not limited to those who perform the functions of contract sterilization, installation, relabeling, remanufacturing, repacking, or specification development, and initial distributors of foreign entities performing these functions.(p) Manufacturing material.指任何用于或用于催化制造过程的任何原料或物质, 在制造过程中发生的陪伴的成份或副产物, 其在制品器械中/上呈现为残留物或杂质, 它不是制造商的设计或意图.(p)Manufacturing material means any material or substance used in or used to facilitate the manufacturing process, a concomitant constituent, or a byproduct constituent produced during the manufacturing process, which is present in or on the finished device as a residue or impurity not by design or intent of the manufacturer.(q)分歧格Nonconformity.未满足规定的要求.(q)Nonconformity means the nonfulfillment of a specified requirement.(r)产物Product.部件、原资料、在制品、制品和返回品. (r)Product means components, manufacturing materials, in- process devices, finished devices, and returned devices.(s)质量Quality.一组固有特性满足要求的法式, 包括平安和性能.(s)Quality means the totality of features andcharacteristics that bear on the ability of a device to satisfy fitness-for-use, including safety and performance.(t)质量审核Quality Audit.按规定的时间间隙和频率, 对制造商的质量体系进行系统、客观的检查, 以确定质量体系活动及其结果符合质量体系法式, 这些法式获得有效执行, 法式适应质量目标的需求.(t)Quality audit means a systematic, independent examination of a manufacturer's quality system that is performed at defined intervals and at sufficient frequency to determine whether both quality system activities and the results of such activities comply with quality system procedures, that these procedures are implemented effectively, and that these procedures are suitable to achieve quality system objectives.(u)质量方针Quality policy.由制造商的最高管理者发布的组织总的质量宗旨和方向.(u)Quality policy means the overall intentions and direction of an organization with respect to quality, as established by management with executive responsibility.(v)质量体系Quality system.质量管理的组织结构、职责、法式、过程和资源.(v)Quality system means the organizational structure, responsibilities, procedures, processes, and resources for implementing quality management.(w) Remanufacturer.指对制品器械进行处理、修整、修复、重新包装、恢复或其它活动的人, 使制品器械的性能、平安规范或预期用途发生重年夜更改.(w)Remanufacturer means any person who processes, conditions, renovates, repackages, restores, or does any other act to a finished device that significantly changes the finished device's performance or safety specifications, or intended use.(x)返工Rework.为使分歧格品在其交付前符合DMR的要求而采用的办法.(x)Rework means action taken on a nonconforming product so that it will fulfill the specified DMR requirements before it is released for distribution.(y)规范Specification.产物、过程、服务或其它活动应符合的要求.(y)Specification means any requirement with which a product, process, service, or other activity must conform.(z)确认Validation.通过检查和提供客观证据证明满足预期用途的要求.(z)Validation means confirmation by examination and provision of objective evidence that the particular requirements for a specific intended use can be consistently fulfilled.(1)过程确认Process validation.根据客观证据确定过程可继续发生满足预先确定例范的结果或产物.(1)Process validation means establishing by objective evidence that a process consistently produces a result or product meeting its predetermined specifications.(2)设计确认Design validation.根据客观证据确定设备规范符合使用者的需求和预期用途.(2)Design validation means establishing by objective evidence that device specifications conform with user needs and intended use(s).(aa)验证Verification.通过检查和提供客观证据证明满足规定的要求.(aa)Verification means confirmation by examination and provision of objective evidence that specified requirements have been fulfilled.Sec.820.5 质量体系/ Quality system. 制造商应建立并实施适应特定的医疗器械设计或制造, 并符合本部份要求的质量体系.Each manufacturer shall establish and maintain a quality system that is appropriate for the specific medical device(s) designed or manufactured, and that meets the requirements of this part.Subpart B——质量体系要求/Quality System Requirements Sec.820.20 管理职责/Management responsibility (a)质量方针:负有执行职责的管理者应建立质量方针和目标以及在质量方面的许诺, 应保证组织内所有级别都能正确理解并执行质量方针.(a)Quality policy.Management with executive responsibility shall establish its policy and objectives for, and commitment to, quality. Management with executive responsibility shall ensure that the qualitypolicy is understood, implemented, and maintained at all levels of the organization.(b)组织:建立并坚持适宜的组织结构, 确保产物的设计和生产符合本部份(Part 820)的要求.(b)Organization.Each manufacturer shall establish and maintain an adequate organizational structure to ensure that devices are designed and produced in accordance with the requirements of this part.(1)职责和权限.制造商应明确影响质量的管理、把持和评价人员的职责、权限及相互关系, 为其提供执行这些工作必需的自主权和权限.(1)Responsibility and authority.Each manufacturer shall establish the appropriate responsibility, authority, and interrelation of all personnel who manage, perform, and assess work affecting quality, and provide the independence and authority necessary to perform these tasks.(2)资源.制造商应提供适当的资源, 包括由经过培训的人员, 执行管理、把持和包括内部质量审核在内的活动, 以符合本部份(Part 820)的要求.2)Resources.Each manufacturer shall provide adequate resources, including the assignment of trained personnel, for management, performance of work, and assessment activities, including internal quality audits, to meet the requirements of this part.(3)管理者代表.最高管理者应在管理层中以书面方式指定一名管理者代表, 无论其在其它方面的职责如何, 应具有以下方面的职责和权限:(3)Management representative.Management with executive responsibility shall appoint, and document such appointment of, a member of management who, irrespective of other responsibilities, shall have established authority over and responsibility for:(i)确保根据本部份(Part 820)的要求有效地建立、实施和坚持质量管理体系;(i) Ensuring that quality system requirements are effectively established and effectively maintained in accordance with this part; and(ii)向负有执行职责的管理者陈说质量体系运行情况, 以供评审.(ii) Reporting on the performance of the quality system to management with executive responsibility for review.(c)管理评审.负有执行职责的管理者, 应按法式规定的时间间隔对证量体系进行审核.确保质量体系的继续适宜性和有效性, 以满足本标准的要求和组织规定的质量方针和目标.评审的日期和结果应形成文件并记录.(c)Management review.Management with executive responsibility shall review the suitability and effectiveness of the quality system at defined intervals and with sufficient frequency according to established procedures to ensure that the quality system satisfies the requirements of this part and the manufacturer's established quality policy and objectives. The dates and results of quality system reviews shall be documented.(d)质量规画.制造商应建立质量规画, 确定设计和制造设备所需的质量准则、资源和活动, 形成质量计划.组织应确定如何满足质量要求.(d)Quality planning. Each manufacturer shall establish a quality plan which defines the quality practices, resources, and activities relevant to devices that aredesigned and manufactured. The manufacturer shall establish how the requirements for quality will be met.(e)质量体系法式.制造商应建立质量体系法式和规范, 适用时应建立质量体系的文件化的结构描述.(e)Quality system procedures.Each manufacturer shall establish quality system procedures and instructions. An outline of the structure of the documentation used in the quality system shall be established where appropriate. Sec.820.22 质量审核Quality audit.制造商应建立并实施质量审核法式和活动, 以确保质量体系符合既定的质量体系要求, 确定质量体系的有效性.质量审核应由与所审核的活动无直接责任的人员进行.纠正办法, 需要时包括对不符合项的重新审核.每次质量审核和重新审核的结果应形成陈说, 陈说要经对审核负有责任的管理者评审.审核和重新审核的日期和结果应予记录.Each manufacturer shall establish procedures for quality audits and conduct such audits to assure that the quality system is in compliance with the established quality system requirements and to determine the effectiveness of the quality system. Quality audits shall be conducted byindividuals who do not have direct responsibility for the matters being audited. Corrective action(s), including a reaudit of deficient matters, shall be taken when necessary. A report of the results of each quality audit, and reaudit(s) where taken, shall be made and such reports shall be reviewed by management having responsibility for the matters audited. The dates and results of quality audits and reaudits shall be documented.Sec.820.25 人员/Personnel (a)概述.制造商应有足够的人员, 经过需要的教育、工作布景、专业培训和相关的经验, 以保证所有法规要求的活动能够获得正确的执行.(a)General.Each manufacturer shall have sufficient personnel with the necessary education, background, training, and experience to assure that all activities required by this part are correctly performed.(b)培训.制造商应建立培训的文件, 明确培训需求, 保证所有人员都能得充沛的培训, 以保证满足工作的要求.培训应形成记录.(b)Training. Each manufacturer shall establish proceduresfor identifying training needs and ensure that all personnel are trained to adequately perform their assigned responsibilities. Training shall be documented.(1)作为培训的一部份, 应使员工意识到他们的特殊工作中的不正确的把持可造成设备的缺陷.(1) As part of their training, personnel shall be made aware of device defects which may occur from the improper performance of their specific jobs.(2)负有验证和确认职责的人员应意识到, 在其工作中会遇到缺陷和毛病.(2) Personnel who perform verification and validation activities shall be made aware of defects and errors that may be encountered as part of their job functions.(1) Each manufacturer of any class III or class II device,and the class I devices listed in paragraph (a)(2) of this section, shall establish and maintain procedures to control the design of the device in order to ensure that specified design requirements are met.(2) The following class I devices are subject to design controls:(i) Devices automated with computer software; and(ii) The devices listed in the following chart.Device868.6810 导管、呼吸机878.4460 手套、外科医生用手套880.6760 阻止、呵护用品892.5650 生化、涂药器、放射性、手工制造(Manual)892.5740 源、放射治疗(b)设计和开发规画.组织应建立并实施设计和开发计划, 其内容描述或包括了设计和开发的相关活动并界说了执行的职责.计划应明确并描述分歧部份/组间的接口及活动, 其结果是设计输入和开发过程.计划应随着设计和开发的推进进行评审、更新, 并经批准.(b)Design and development planning.Each manufacturer shall establish and maintain plans that describe or reference the design and development activities and define responsibility for implementation. The plans shall identify and describe the interfaces with different groups or activities that provide, or result in, input to the design and development process. The plans shall be reviewed, updated, and approved as design and development evolves.(c)设计输入.组织应建立并坚持法式, 以保证与产物相关的设计要求是适宜的, 并满足设备的预期用途, 包括使用者和患者的需要.这个法式应包括解决任何不完全、不明确和相互矛盾的要求的机制.设计输入要求应经审核, 并经指定的人员审核和批准.审批应包括审批人员的签名和日期, 审批应予记录.(c)Design input.Each manufacturer shall establish and maintain procedures to ensure that the design requirements relating to a device are appropriate and address the intended use of the device, including the needs of the user and patient. The procedures shall include a mechanism for addressing incomplete, ambiguous, or conflicting requirements. The design input requirements shall be documented and shall be reviewed and approved by a designated individual(s). The approval, including the date and signature of the individual(s) approving the requirements, shall be documented.(d)设计输出.组织应建立并坚持文件化的设计输出法式, 使经过评审的设计输出文件满足设计输入的要求.设计输出法式应包括或涉及接收标准, 确保实现设备基本的、适用的功能.设计输出应是文件化的, 在发布前应经评审和批准.审批应文件化, 包括批准人的签名及日期.(d)Design output.Each manufacturer shall establish and maintain procedures for defining and documenting design output in terms that allow an adequate evaluation of conformance to design input requirements. Design output procedures shall contain or make reference to acceptance criteria and shall ensure that those design outputs that are essential for the proper functioning of the device are identified. Design output shall be documented, reviewed, and approved before release. The approval, including the date and signature of the individual(s) approving the output, shall be documented.(e)设计评审.组织应建立并实施法式, 确保在产物设计开发的适当阶段, 有计划地对设计结果进行正式的评审.法式应确保每次设计评审的介入者, 应包括与被评审的设计阶段有关的所有职能部份的代表, 和一名或多名与被评审设计阶段无直接责任的人员, 需要时也可包括其它专家.评审结果, 包括设计标识(identification of design)、日期、评审的人员, 应在设计历史文件中予以记录.(e)Design review.Each manufacturer shall establish and maintain procedures to ensure that formal documented reviews of the design results are planned and conductedat appropriate stages of the device's design development. The procedures shall ensure that participants at each design review include representatives of all functions concerned with the design stage being reviewed and an individual(s) who does not have direct responsibility for the design stage being reviewed, as well as any specialists needed. The results of a design review, including identification of the design, the date, and the individual(s) performing the review, shall be documented in the design history file (the DHF).(f)设计验证.组织应建立并实施设计验证的法式.确保设计输出满足设计输入的要求.设计验证的结果, 包括设计标识(identification of design)、方法、日期、验证的人员, 应在设计历史文件中予以记录.(f)Design verification. Each manufacturer shall establish and maintain procedures for verifying the device design. Design verification shall confirm that the design output meets the design input requirements. The results of the design verification, including identification of the design, method(s), the date, and the individual(s) performing the verification, shall be documented in theDHF.(g)设计确认.组织应建立并实施设计确认法式.设计确认应在规定的把持条件下, 对最初的产物、批次或其等价物上进行.设计确认应确保产物满足规定的用户需求和预期的使用要求, 也包括在实际或模拟的使用条件下对产物单位进行试验.适用时, 设计确认应包括软件确认和风险分析.设计确认的结果, 包括设计标识(identification of design)、方法、日期、确认的人员, 应在设计历史文件中予以记录.(g)Design validation.Each manufacturer shall establish and maintain procedures for validating the device design. Design validation shall be performed under defined operating conditions on initial production units, lots, or batches, or their equivalents. Design validation shall ensure that devices conform to defined user needs and intended uses and shall include testing of production units under actual or simulated use conditions. Design validation shall include software validation and risk analysis, where appropriate. The results of the design validation, including identification of the design, method(s), the date, and the individual(s) performing the validation, shall be documented in the DHF.(h)设计转换.组织应建立并坚持文件化的法式, 以保证产物的设计能够正确的转换成产物的规范.(h)Design transfer. Each manufacturer shall establish and maintain procedures to ensure that the device design is correctly translated into production specifications.(i)设计更改.组织应建立并坚持法式, 在执行前设计更改应被识别、文件化、确认或适用时经验证、评审和批准.(i)Design changes. Each manufacturer shall establish and maintain procedures for the identification, documentation, validation or where appropriate verification, review, and approval of design changes before their implementation.(j)设计历史文件.组织应建立并坚持每个型号的产物的DHF.DHF应包括或涉及需要的记录, 以证明设计的进程符合被批准的设计计划和本部份的要求.(j)Design history file. Each manufacturer shall establish and maintain a DHF for each type of device. The DHF shall contain or reference the records necessary to demonstrate that the design was developed in accordance with the approved design plan and the requirements of this part.Subpart D——文件控制Document ControlsSec.820.40 文件控制Document Controls 组织应建立并实施法式, 以控制所有本部份要求的文件.法式应包括:Each manufacturer shall establish and maintain procedures to control all documents that are required by this part. The procedures shall provide for the following:(a)文件的批准和发布.所有文件在发布前应由授权人员评审、批准其适宜性, 以满足本部份的要求.文件的批准, 包括批准发布人员的签名及日期应形成记录.确保在文件适用的场所能够获得相关文件, 从所有发放或使用场所及时撤出作废文件, 以防止作废文件的非预期使用.(a)Document approval and distribution.Each manufacturer shall designate an individual(s) to review for adequacy and approve prior to issuance all documents established to meet the requirements of this part. The approval, including the date and signature of the individual(s) approving the document, shall be documented. Documents established to meet the requirements of this part shall be available at all locations for which they are designated, used, or otherwise necessary, and all obsolete documents shall be promptly removed from all。
FDAQSR820教材(康志华)解析

章节A 概述--820.3 定义
(e) 设计历史文件Design history file(DHF)。 最终产品的设计历史记录的汇总。
(b) 部件Component。
所有成为最终、包装、标识的设备的一部分的原 材料、物资、件、部分、软件、连接件、标签或 它们的集合。
(c) 控制号Control number。
唯一性标识,如由字母、数字或它们的组合形成 的唯一性组合,由控制号可以确定一批或一个设 备的制造、包装、标签和交付的历史。
QSR820 框架
H—检验活动 I— 不合格品 J—纠正与预防措施 K—标贴和包装控制 L—搬运、贮存、交付与安装 M—记录 N—服务 O—统计技术
章节A 概述--820.1 范围
820.1 范围
适用性
本质量体系法规阐明了当前优良制造过程(CGMP)的 要求。本标准适用于所有应用于人类的最终产品的设计、 制造、包装、标识、贮存、安装和服务,所适用的管理 方法、设备和控制。
FDA QSR820培训
康志华 2014年8月30日
目录
一、FDA、QSR820简介 二、QSR820质量体系法规
一、FDA、QSR 820简介
FDA:美国食品、药品监督管理局 21CFR820是根据相关条款授要而制定的规
范医疗器械企业质量体系法规,即Quality System Regulation,简称QSR或QSR820.
章节A 概述--820.1 范围ห้องสมุดไป่ตู้
限制。 除非特别规定,则本部分质量体系法规是本章其它部分 法规的补充要求。在不能符合所有适用的法规,包括本 章此部分和其它部分的情况,特别是对讨论中的设备, 此法规应取代其它通用要求。
权限。
美国FDA-医疗器械体系法规QSR820中英文版2015.06

培训教材美国FDA 医疗器械体系法规QSR820中文版2015.06Part 820——质量体系法规——目录Subpart A- 总则820.1 范围820.3 定义820.5 质量体系Subpart B –质量体系要求820.20 管理职责820.22 质量审核820.25 人员Subpart C- 设计控制820.30 设计控制Subpart D- 文件控制820.40 文件控制Subpart E- 采购控制820.50 采购控制Subpart F- 标识与可追溯性820.60 标识820.65 可追溯性Subpart G - 生产和过程控制820.70 生产和过程控制820.72 检验、测量和试验设备820.75 过程确认Subpart H - 验收活动:820.80 进货、过程和成品器械检验820.86 检验状态Subpart I –不合格品820.90 不合格品Subpart J - 纠正和预防措施820.100 纠正和预防措施Subpart K –标识和包装控制820.120 设备标签820.130 设备包装Subpart L –搬运/储存/分销和安装820.140 搬运820.150 贮存820.160 分销820.170 安装Subpart L –记录820.180 记录的通用要求820.181 设备主要记录820.184 设备历史记录820.186 质量体系记录820.198 投诉文件Subpart M –服务820.200 服务Subpart N –统计技术820.250 统计技术Subpart A——总则Subpart A--General ProvisionsSec.820.1 范围Sec. 820.1 Scope.(a)适用性Applicability。
(1)本质量体系法规阐明了当前良好制造法规Current good manufacturing practice (CGMP)的要求。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
美国的医疗器械法规体系--QSR820 美国食品药品监督管理局(FDA)是负责医疗器械管理的政府机构。
其根据各相关法律授权而制定的各类法规性的文件编号为21CFRxxxx(xxxx为阿拉伯数字)。
其中21CFR820是FDA根据《联邦食品,药品和化妆品法案》第501, 502, 510, 513, 514, 515, 518, 519, 520, 522, 701, 704, 801, 803条款的授权而制定的规范医疗器械企业质量体系要求的法规,即Quality System Regulation,简称QSR或QSR820。
谁要遵守QSR820?
21QSR820。
1规定,所有在美国和波多黎各境内的,或者有产品出口到美国和波多黎各境内的医疗器械企业必须按QSR820的要求建立质量体系。
各企业可以根据实际情况,满足QSR中与自己活动相关的条款。
QSR820不适用于医疗器械零件生产商,但FDA鼓励这类企业以QSR820中适用的条款为指导。
QSR820不适用于人血和血制品生产商。
这类企业应遵循21CFR606的规定。
谁来检查企业是否符合QSR820?
FDA下属的CDRH(器械与放射健康中心)是专职负责医疗器械企业管理的政府机构,其根据FDA的授权安排检查员到个企业进行工厂检查。
对美国境内企业一般每两年检查一次,境外企业不定期检查。
所有检查费用由FDA承担。
在欧洲和美国本土,FDA也授权TUV等第三方机构进行工厂检查,扥企业要支付相应费用。
无论谁来检查,都只是一个符合性检查,不颁发任何证书,不属于【信息咨询】活动。
如何检查企业是否符合QSR820?
QSIT(质量体系检查技术)是FDA检查员的必修课程,也是FDA Quality Systems Inspection Reengineering Team专门编制的检查员手册。
该文件详细介绍了检查方法,关注点,无论对FDA检查员还是企业内审员/供应商审核员都具有参考价值。
QSR820概况
现行版本的QSR820颁布于1996年10月7日,正式生效于1997年6月1日,亦被称为美国医疗器械行业的现行良好的规范(Current Good Manufacture Practice,简称cGMP)。
全文一共15个章节:
A总则
B质量体系要求
C设计控制
D文件控制
E采购控制
F识别与可追溯性
G生产于过程控制
H验收活动
I不合格产品
J纠正与预防措施
K标签与包装控制
L搬运,存储,发运与安装
M记录
N服务
O统计技术
总体而言,这是一套在结构上不同于IS013485:2003,要求上与IS013485:2003基本相同,规定上更加明确的质量管理体系法规:
(括号内为IS013485:2003对应条款)
B: 质量体系要求(4.1, 5, 6.2, 8.2.2)
C: 设计控制(7.3)
D: 文件控制(4.2)
E: 采购控制(7.4)
F: 识别与可追溯性(7.5.3)
G: 生产与过程控制(7.5, 6.3, 6.4, 7.6)
H: 验收活动(8.2.4)
I: 不合格产品(8.3)
J: 纠正与预防措施(8.5.2, 8.5.3)
K:标签与包装控制
L:搬运、存储、发运与安装(7.5.5, 7.5.1) M:记录(4.2, 8.5.1)
N:服务(7.5.1)
O:统计技术(8.1)。