ELISA(酶联免疫吸附测定)实验报告
酶联免疫吸附测定实验报告

酶联免疫吸附测定实验报告酶联免疫吸附测定实验报告酶联免疫吸附测定(Enzyme-Linked Immunosorbent Assay, ELISA)是一种常用的实验技术,用于检测和定量分析生物样本中特定分子的存在。
本实验旨在通过ELISA技术,对特定抗原在样本中的含量进行测定,并了解其原理及应用。
一、实验原理ELISA技术是一种基于免疫反应的实验方法,其原理主要包括固相吸附、特异性结合、酶标记和显色反应。
首先,在微孔板中固定特异性抗体,形成固相吸附。
然后,加入待测样本,样本中的目标抗原与固相抗体结合。
接着,加入酶标记的二抗与目标抗原结合,形成特异性结合。
最后,通过添加显色底物,酶催化反应产生可见的颜色变化,从而定量分析目标抗原的含量。
二、实验步骤1. 准备样本和试剂:收集待测样本,如血清、尿液或细胞培养上清液,并准备好ELISA试剂盒中的各种试剂。
2. 板洗涤:将微孔板放入洗板机中,加入洗板缓冲液,进行洗涤步骤,以去除未结合的物质。
3. 固相吸附:将特异性抗体加入微孔板孔中,孵育一段时间,使其与固相吸附。
4. 样本孵育:将待测样本加入各孔中,与固相抗体结合,在恒温条件下孵育。
5. 二抗结合:加入酶标记的二抗,与目标抗原结合形成特异性结合。
6. 洗涤:再次进行洗板步骤,去除未结合的物质。
7. 底物反应:加入显色底物,酶催化反应产生可见的颜色变化。
8. 反应终止:加入终止液,停止酶催化反应。
9. 测定吸光度:使用酶标仪测定各孔的吸光度值。
10. 数据分析:根据吸光度值,绘制标准曲线,通过比较待测样本的吸光度值,计算出目标抗原的浓度。
三、实验结果通过ELISA实验,我们成功测定了待测样本中目标抗原的含量。
根据标准曲线,我们计算出了各样本中目标抗原的浓度。
这些结果将有助于我们了解样本中特定分子的水平,从而进一步研究其生物学功能和相关疾病的发生机制。
四、实验应用ELISA技术具有广泛的应用领域,包括生物医学研究、临床诊断、药物开发等。
酶联免疫吸附试验实验报告

酶联免疫吸附试验实验报告一、实验目的。
本实验旨在通过酶联免疫吸附试验(ELISA)检测目标蛋白或抗原的存在与浓度,以及对特定抗体的识别和结合情况。
通过本实验,我们可以了解到酶联免疫吸附试验的原理和操作步骤,以及在生物医学研究和临床诊断中的应用。
二、实验原理。
酶联免疫吸附试验是一种利用酶和抗体的特异性结合来检测抗原或抗体的方法。
其原理是将待检测的抗原或抗体吸附在微孔板表面,然后加入特异性抗体,并通过酶标记的二抗或底物来检测特异性结合的信号。
当待测物与特异性抗体结合后,通过底物的酶反应产生可定量的颜色反应,从而测定待测物的浓度。
三、实验材料和方法。
1. 实验材料,微孔板、待测抗原或抗体、特异性抗体、酶标记的二抗、底物溶液等。
2. 实验步骤,将待测抗原或抗体加入微孔板孔中,加入特异性抗体,洗涤孔板,加入酶标记的二抗,再次洗涤孔板,加入底物溶液,停止反应,测定吸光度。
四、实验结果。
通过本次实验,我们成功检测出待测物的存在与浓度,并观察到特异性抗体与待测物的结合情况。
根据实验结果,我们可以得出待测物的浓度,并进一步分析其在生物学过程中的作用和意义。
五、实验结论。
酶联免疫吸附试验是一种高度敏感和特异性的实验方法,广泛应用于生物医学研究和临床诊断领域。
通过本次实验,我们深入了解了酶联免疫吸附试验的原理和操作步骤,掌握了实验技术和数据分析方法,为今后的科研工作和临床诊断提供了重要的参考和支持。
六、实验展望。
酶联免疫吸附试验作为一种重要的生物学实验方法,具有广阔的应用前景。
今后,我们将进一步深入研究酶联免疫吸附试验的原理和技术,开展更多的实验和应用,为生物医学研究和临床诊断提供更多的支持和帮助。
七、参考文献。
1. Smith, J. et al. (2010). Enzyme-linked immunosorbent assay (ELISA). Methods Mol Biol, 588, 89-94.2. Green, N. M. (2015). Avidin and streptavidin. Methods Enzymol, 184, 51-67.以上就是本次酶联免疫吸附试验的实验报告,谢谢阅读。
酶联免疫吸附实验报告

酶联免疫吸附实验报告酶联免疫吸附实验报告酶联免疫吸附实验(enzyme-linked immunosorbent assay, ELISA)是一种常用的生物化学技术,用于检测和定量分析特定抗原或抗体的存在。
该实验结合了免疫学和酶学的原理,通过酶的催化作用来实现对目标物的检测。
本报告将介绍酶联免疫吸附实验的基本原理、操作步骤以及结果分析。
一、实验原理酶联免疫吸附实验的原理基于免疫学的特异性识别和酶学的催化作用。
实验中,首先将待测物(抗原或抗体)与特异性的抗体或抗原结合,形成抗原-抗体复合物。
然后,将该复合物与与酶标记的抗体或抗原结合,形成二抗或二抗原复合物。
最后,通过添加底物,利用酶的催化作用产生可测量的信号,从而确定待测物的存在或浓度。
二、实验步骤1. 准备样品和试剂:收集待测物样品,并准备所需的试剂,如抗体、底物等。
2. 预处理样品:根据待测物的性质,进行必要的样品预处理,如稀释、去除干扰物等。
3. 涂布固相支持物:将特异性抗体或抗原溶液均匀涂布在固相支持物(如酶标板)上,使其吸附。
4. 孵育:将待测物样品加入酶标板中,与固相支持物上的抗体或抗原结合,形成复合物。
然后加入酶标记的抗体或抗原,形成二抗或二抗原复合物。
孵育一段时间,以便复合物的形成。
5. 洗涤:将酶标板反复洗涤,去除未结合的物质。
6. 底物反应:加入适当的底物,使底物与酶发生反应,产生可测量的信号。
底物的选择与酶的选择有关,常用的底物有TMB(3,3',5,5'-四甲基联苯胺)等。
7. 反应停止:通过添加反应停止剂,停止底物的反应,并使产生的信号停止发展。
8. 读取结果:使用酶标仪或其他适当的仪器,测量底物反应产生的信号的强度。
根据标准曲线或对照样品,确定待测物的浓度或存在与否。
三、结果分析根据实验结果,可以定量测定待测物的浓度或判断其存在与否。
一般来说,酶标仪测得的信号强度与待测物的浓度成正比,可以通过标准曲线来确定待测物的浓度。
elisa实验报告范文

elisa实验报告范文篇一:Elia实验名称:酶联免疫吸附剂测定实验原理:酶联免疫吸附测定是一种免疫测定技术。
测定中,先使抗原吸附在固相载体上,然后加待测的抗体,再用某种酶标记抗体,形成抗原-抗体-酶标记抗体的“双抗体夹心”,此时酶仍保有活性,同时标记抗体亦有免疫活性。
之后再加入酶的底物,在酶的催化下产生反应并产生有色物,颜色深浅与待测物质的量直接相关。
至此,酶的催化放大作用与免疫反应的特异性相完美结合,提高了测定的准确性与灵敏度。
实验材料与试剂:1.聚苯乙烯微量细胞培养板(平板,96孔)。
2.酶联免疫检测仪。
3.辣根过氧化物酶羊抗兔IgG。
4.包被液:0.05mol/L碳酸缓冲液(pH9.6):Na2CO30.15g,NaHCO30.293g,蒸馏水稀释至100ml。
5.稀释液(PBS-Tween):NaCl8g,KCl0.2g,KH2PO40.2g,Na2HPO4·12H2O2.9g,Tween-20,0.5ml,蒸馏水加至1000ml。
6.洗涤液:同稀释液。
7.封闭液:0.5%(质量分数)BSA(用PBS配制)。
8.邻苯二胺溶液(底物):配制:0.1mol/L柠檬酸(2.1g/100ml),取6.1ml,0.2mol/LNa2HPO4·12H2O(7.163g/100ml),取6.4ml,加蒸馏水12.5ml,取邻苯二胺8mg(溶解);临用前加30%(体积分数)H2O240μl。
9.终止液:2mol/LH2SO4。
实验步骤:1.包被抗原:用包被液将抗原作适当稀释,一般为1~10μg/孔,每孔加200μl,37℃温育30min。
2.洗涤:倒尽板孔中液体,加200μl洗涤液,反复三次,最后将反应板倒置在吸水纸上,使孔中洗涤液流尽。
3.加封闭液200μl,37℃温育30min。
4.洗涤同2。
5.加被检血清:用稀释液将被检血清作几种稀释,每孔200μl。
同时作稀释液对照。
37℃温育30min。
elisa检测实验报告

elisa检测实验报告ELISA 检测实验报告一、实验目的本次 ELISA 检测实验的目的是定量检测样本中特定抗原或抗体的浓度,以评估实验对象的生理或病理状态。
二、实验原理ELISA(酶联免疫吸附测定)是一种基于抗原抗体特异性结合反应的免疫测定技术。
其基本原理是将已知的抗原或抗体固定在固相载体(如聚苯乙烯微孔板)表面,然后加入待检样本,样本中的待测抗原或抗体与固相载体上的抗原或抗体发生特异性结合。
接着,加入酶标记的第二抗体(或抗原),形成抗原抗体酶标记抗体复合物。
最后,加入底物,酶催化底物反应生成有色产物,通过测定有色产物的吸光度值,即可定量分析样本中待测抗原或抗体的浓度。
三、实验材料与设备1、试剂包被缓冲液(碳酸盐缓冲液,pH 96)封闭液(含 1% 5% 牛血清白蛋白的 PBS 缓冲液)洗涤液(含 005% Tween-20 的 PBS 缓冲液)样本稀释液(PBS 缓冲液)标准品(已知浓度的抗原或抗体)酶标记的二抗(辣根过氧化物酶或碱性磷酸酶标记)底物溶液(如 TMB 或 pNPP)终止液(如 2M 硫酸或 1M 氢氧化钠)2、仪器与设备酶标仪(能够读取 450nm 或 492nm 波长的吸光度值)恒温培养箱移液器(量程分别为20μL、200μL、1000μL)聚苯乙烯微孔板离心机四、实验步骤1、包被将已知浓度的抗原或抗体用包被缓冲液稀释至适当浓度。
向聚苯乙烯微孔板的每孔中加入100μL 稀释后的包被液,4℃过夜或 37℃孵育 2 3 小时。
2、封闭倒掉包被液,用洗涤液洗涤微孔板 3 次,每次浸泡 3 分钟,然后拍干。
每孔加入200μL 封闭液,37℃孵育 1 2 小时。
3、加样倒掉封闭液,用洗涤液洗涤微孔板 3 次,然后拍干。
将待检样本和标准品用样本稀释液进行梯度稀释。
向微孔板的每孔中分别加入100μL 稀释后的样本和标准品,空白对照孔加入100μL 样本稀释液。
37℃孵育 1 2 小时。
ELISA酶联免疫吸附试验报告
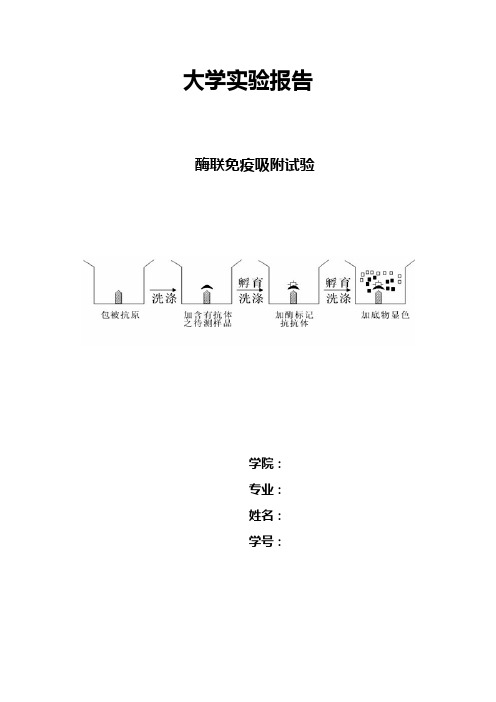
大学实验报告酶联免疫吸附试验学院:专业:姓名:学号:一.实验原理酶联免疫吸附试验(ELISA)是一种用酶标记抗原或抗体的方法,此法是将抗原、抗体免疫反应的特异性和酶的高效催化作用原理有机地结合起来的技术,可敏感地检测体液中微量的特异性抗原或抗体。
基本原理如下:①先使用抗原或抗体结合到某种固相载体表面,并保持其免疫活性;②使抗原或抗体与某种酶联结成酶标抗原或酶标抗体,这种酶标抗原或酶标抗体即保留了其免疫活性,又保留了酶活性;③试验时,把受检标本(抗体或抗原)和酶标抗原或酶标抗体按不同的步骤与固相载体表面的抗原或抗体起反应,用洗涤法去除固相载体上的游离物质,最后结合在固相载体上的酶量与标本中受检物质的多少有关。
当加入酶反应的底物后,底物被酶催化而产生有色物质。
颜色反应深浅与受检抗原或抗体的量成正比。
因此,可借助颜色反应的深浅来定性、定量抗体或抗原。
本技术的特点是敏感性高,特异性强,操作简易,结果容易观察。
图1 ELISA实验原理示意图二.实验材料BSA抗原、兔抗BSA抗体(一抗,分别用PBS稀释成1:2, 1:4, 1:16, 1:32, 1:64, 1:128, 1:256七个浓度)、酶标羊抗兔IgG(二抗)、0.05M PH9.6碳酸盐包被缓冲液、洗涤液(PBS)、底物溶液、2mol/L H2SO4。
三.实验方法1.包被板的处理:加BSA(包被缓冲液稀释为10μg/ml)至酶标板中,每孔100μl,置湿盒中4℃过夜。
第二天取出,用PBS洗涤3-4次。
2.取包被好的酶标板,在第一孔中加入100μl PBS作阴性对照,在第2-8孔加入上述稀释的一抗各100μl,置37℃保温1h。
3.倾去液体,用PBS洗涤3-4次后加入二抗各100μl,置37℃保温30min。
4.倾去液体,用PBS洗涤3-4次后加入底物溶液各100μl,在暗处37℃避光显色20min,最后加入50μl、2M H2SO4终止反应。
5.在波长490nm处,测定各孔的光密度吸收值。
酶联免疫吸附试验实验报告

酶联免疫吸附试验实验报告酶联免疫吸附试验实验报告酶联免疫吸附试验(enzyme-linked immunosorbent assay,ELISA)是一种常用的生物化学实验技术,用于检测和定量分析抗原或抗体的存在与浓度。
本实验旨在通过ELISA技术,检测和定量分析一种特定抗原在不同样本中的浓度变化。
实验材料和方法:1. 样本制备:收集不同来源的样本,如血清、尿液等,并进行离心分离获得上清液。
2. 酶标板涂覆:将待测抗原溶液加入酶标板孔中,保持一定的温度和时间,使抗原均匀地附着在酶标板表面。
3. 阻断:加入阻断液,封闭未被抗原占据的酶标板孔,避免非特异性结合。
4. 抗体结合:加入特异性抗体,与抗原结合形成抗原-抗体复合物。
5. 酶标记抗体结合:加入酶标记抗体,与抗原-抗体复合物结合,形成二抗夹心复合物。
6. 显色反应:加入底物,使酶标记抗体催化底物发生颜色变化。
7. 反应停止:加入停止液,终止显色反应。
8. 测定吸光度:使用酶标仪测定酶标板孔中的吸光度。
实验结果:根据测定吸光度值,我们可以得到不同样本中抗原的浓度变化。
通过比较不同样本的吸光度值,可以了解抗原在不同条件下的表达情况以及不同样本中抗原的相对浓度。
实验讨论:ELISA技术在生物医学领域具有广泛的应用,尤其在疾病诊断和药物研发方面起到了重要的作用。
通过ELISA技术,可以检测和定量分析目标抗原的存在与浓度,从而判断疾病的发生与发展,为临床诊断提供依据。
在本次实验中,我们选择了特定抗原作为目标,通过ELISA技术对其进行检测和定量分析。
实验结果显示,不同样本中抗原的浓度存在差异,这可能与样本来源、处理方法等因素有关。
通过进一步的研究和分析,可以进一步探究这些差异的原因,并为临床诊断和治疗提供更准确的依据。
此外,ELISA技术还可以用于药物研发过程中的药物筛选和药效评价。
通过检测药物对特定抗原的影响,可以评估药物的疗效和安全性,为新药的研发提供重要的参考依据。
ELISA实验报告

ELISA实验报告实验目的:本实验旨在通过ELISA(酶联免疫吸附试验)技术来检测细胞培养上清液中的蛋白质含量,从而了解该细胞株的蛋白质表达水平,进一步研究相关的细胞分子机制。
实验原理:酶联免疫吸附试验是一种常用的免疫学实验方法,通过特异性抗体与待测物结合来实现对特定蛋白质的检测。
该实验主要分为三个步骤:包被抗原、特异性抗体结合和酶底物显色。
实验材料:1.待测细胞上清液2.包被抗原3.特异性抗体4.酶标记的二抗5.酶底物6.缓冲液7.清洗缓冲液8.吸光度测定仪实验步骤:1.包被抗原的处理:将抗原溶液加入微孔板孔中,使其均匀附着在孔壁上。
然后将孔中的液体弃去,用清洗缓冲液进行孔的清洗。
2.探针的结合:将待测样品加入已经包被抗原的孔中,使其与抗原结合。
然后将孔中的液体弃去,用清洗缓冲液进行孔的清洗。
3.酶标记的二抗结合:将酶标记的二抗加入已经含有特异性抗体的孔中,使其与特异性抗体结合。
然后将孔中的液体弃去,用清洗缓冲液进行孔的清洗。
4.酶底物加入:将酶底物加入到孔中,使其在酶的作用下发生显色反应。
5.吸光度测定:使用吸光度测定仪读取吸光度值,根据吸光度值可以推断待测样品中蛋白质的含量。
实验结果:经过ELISA实验,我们得到了待测细胞上清液中蛋白质的含量。
根据吸光度值和标准曲线的对照,我们可以计算出待测样品中蛋白质的浓度。
实验结论:实验分析:本次实验利用ELISA技术成功检测了待测细胞上清液中的蛋白质含量。
该方法具有高灵敏度、高特异性、操作简单等优点,可以在生物医学、生物工程等领域广泛应用。
但需要注意的是,ELISA实验在操作过程中需要严格控制实验条件,避免交叉污染和误差的产生。
实验改进:为了进一步提高实验的准确性和可靠性,可以进行以下改进:1.增加重复次数,提高数据的可靠性和稳定性;2.使用更加准确的仪器和试剂;3.对实验流程进行优化,减少操作的差异性。
总结:本次实验通过ELISA技术成功检测了待测细胞上清液中的蛋白质含量,得出了蛋白质表达水平较高的结论。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
ELISALin Chengyu Bio 04 2010030007Experiment Date: 2012-03-12 Submitting Date: 2012-03-211Introduction1.1Background informationELISA (Enzyme-linked Immunosorbent Assay) is a solid-phase assay for antibodiesemploying ligands labeled with enzymes which is widely used for immunological assays.This technique can be applied to detect antigens or antibodies for qualitative orquantitative purpose. Since enzyme reactions are very well known amplificationprocesses, the signal is generated by enzymes which are linked to the detection reagentsin fixed proportions to allow accurate quantification.11.2Major principlesFigure 1 Schematic diagram of ELISA2Figure 2 Procedure of indirect ELISA3As shown in Figure 1 & 2, the general procedure of indirect ELSIA is to: incubate theplate well with antigen, wash off unbounded antigen, incubate with 1st antibody, washoff unbounded 1st antibody, incubate with labeled 2nd antibody, wash off unbounded 2ndantibody, incubate with enzyme substrate solution, and detect optical density or otherindex showing enzyme activity.2Experiment Operation2.1Antigen coating(1)Prepare an antigen solution in coating buffer (human IgG at 0.025mg/ml);(2)Pipette 200 μl antigen solution to each well (Row: B~G; Column: 2~10; Column 11is negative control without antigen) of the microtiter plate;(3)Incubate the plate at 37 ℃for 30 min;(4)Remove the antigen solution;(5)Wash each well with 200 μl with PBS-T for 3 times;(6)Block each well (Row: B~G; Column: 2~11) with 200 μl 0.5% BSA-PBS, andincubate the plate at 37 ℃for 30 min;(7)Remove the blocking solution;(8)Wash each well with 200 μl with PBS-T for 3 times.2.2Primary antibody reaction(1)Dilute the primary antibody (rabbit-anti-human IgG antiserum) in PBS-T fordifferent dilution (from 1:400 to 1:51,200 in 2-folds dilution);(2)Add 200 μl diluted antibody solution to each well following Table 1;Table 1 Scheme to add primary antibody(3)Incubate the plate at 37 ℃for 1 hour;(4)Remove the primary antibody solution;(5)Wash each well with 200 μl PBS-T for 3 times.2.3Application of secondary antibody(1)Dilute the peroxidase conjugated secondary antibody (Goat-anti-rabbit IgG-HRP) inPBS-T at the dilution of 1:20,000 and 1:40,000;(2)Add 200 μl secondary antibody solution to each well following Table 2;(3)Incubate the plate at 37 ℃for 1 hour;(4)Remove the secondary antibody solution;(5)Wash each well with 200 μl PBS-T for 3 times.2.4Substrate development(1)Add 200 μl substrate solution to each well (Row: B~G ,Column: 2~11);(2)Incubate for approximately 3 min;(3)Add 50 μl 2 M H2SO4 to each well to terminate the reaction;(4)Measure optical density at 490 nm.3Raw data and its processing3.1Raw data3.2Data processingSet Row B, C, and D as Group I, and Row E, F, and G as Group II. The processed datais shown in Table 4Table 4 Processed data: optical density of each groupSet different dilutions of primary antibody as x axis, optical density as y axis, drawFigure 3 to illustrate their relation.Figure 3 Relationship between optical density and dilutions of primary antibodyFor the reason that the curve cannot illustrate the relationship enough, change the x axis to nature logarithm of different dilutions of primary antibody. See Figure 4:Figure 4 Relationship between optical density and natural logarithm of dilutions of primary antibodyUsing linear fit for each group, we can figure out that two lines are approximately parallel.In the black curve in Figure 3, there is an oblivious point of inflection which corresponds with the dilution of 1:800. The curve after this point becomes flat, which indicates that the binding between antigens and primary antibodies is saturated in the dilution of 1:800 and higher. This data can suggest that in other immunoenzymatic experiment, the proper dilution of primary antibody will be around, and no higher than 1:800.What’s more, from the red line in Figure 4 we can figure out that the optical density hasa linear relation with natural logarithm of dilutions of the primary antibody.As for comparison between Group I and Group II, from Figure 3 we can figure out thatthe point of infection of blue curve, which corresponds with the dilution of 1:40,000, ison the left, about 1:1600.In Figure 4, the green line (1:40,000) is positioned lower than the red line (1:20,000),which is easy to understand. Lower concentration of secondary antibody means lessbinding with primary antibody during application of secondary antibody.4Results and discussion4.1Results(1)The optical density has an approximately linear relation with the natural logarithmof the dilutions of the primary antibody;(2)For secondary antibody in the dilution of 1:20,000, the proper dilution of primaryantibody is 1:800; for secondary antibody in the dilution of 1:40,000, primaryantibody is recommended to be 1:1600;(3)With the same dilution of antigen and primary antibody, higher concentration ofsecondary antibody will get a higher optical density;4.2Discussion(1)What is the significance of the negative control groups?I.The no primary antibody groups proved that there is no specific bindingbetween antigen and secondary antibody, and provided a background ofnon-specific binding between secondary antibody and antigen;II.The no antigen groups can provide a background of non-specific binding between primary antibody and BSA.(2)Why washing step is essential?Washing each well with PBS-T, which contains tween-20 as detergent, can wash offunbounded antigens and antibodies, including those non-specifically binding. Ifwashing step is omitted, the background index will be higher, and might causeinterference to the result.(3)Why blocking step is essential?After the antigen coating step, the surface of the well is not covered by antigenentirely, i.e. there is still some site leaving blank, which allows other proteins bindto them. Blocking step is to block those blank sites with non-specific bindingmaterial that will not cause interference to the experiment. Thus, the primaryantibody will only bind to the antigen coated in the first step, rather than coat on thesurface as well.(4)What’s the advantage of indirect ELISA comparing with direct ELISA?I.Indirect ELISA can amplify the optical density which we measure.Compared to direct ELISA, the number of secondary antibody binding tothe primary antibody is way larger than the number of primary antibodybinding to the antigen. Thus, optical density will be higher and easier tomeasure, which means a lower error;II.The secondary antibody contains HRP, which is essential for substrate development. Compared to direct ELISA, indirect ELISA need only onekind of antibody contains HRP to perform many kinds of experiment, ratherthan one antibody linked to enzyme for one experiment, which isinconvenient.5Reference【1】/wiki/ELISA【2】/post/9314400054【3】/indirect_elisa。