UPLC法测定奈韦拉平的含量及有关物质
UPLC法测定生长抑素原料药的含量

2 h eodMeia Sh o o az o nvri;3 C lg L eS i eo L nhuU i ri TeS cn dcl c olfL nhuU i sy ol eo i c n azo n esy e t e f f e f c v t
A sr c O jcie T s bit a b t a t bet : oet lha a i, cua n sie h dt dt mie t a cui l v a s r e e t me o e a vp m c
[ 摘 要] 目的 : 建立快速 、 准确 、 灵敏 的测定生 长抑素原料 药的方法。方法: 采用 沃特世 A Q I Y 高液 CUT 超
相 色谱 系统 (P C , U L ) 以磷 酸 ( 取磷 酸 l m , 水 9 0 L 用三 乙胺调 p 1 L加 0 , m H至 2 3 用水稀释至刻度 100m ., 0 L处)
9 .% . o cu i n UPL t o , ih i a i , c u a e s n i v n t o d r p aa i t , o l e vc o 9 1 C n ls : o C me h d wh c srp d a c t , e s ie a d wi g o e e tb l y c u d s r ie f r r t h i
b fe ou in wa s b l h s a d a e o i i sB, r d e t l t n wa s d wi o r t f . / n u r lt s s o a mo i p a eA n c t n t l a g a i n u i s e t f w ae o 3 mL mi ; e re e o u hl O
高效液相色谱法测定奈韦拉平的血药浓度

高效液相色谱法测定奈韦拉平的血药浓度【关键词】色谱法,高压液相;奈韦拉平;血药浓度【关键词】色谱法,高压液相;奈韦拉平;血药浓度奈韦拉平(nevirapine)是人体免疫缺陷病毒(HIV1)的非核苷类逆转录酶抑制剂(NNRTI). 奈韦拉平与HIV1的逆转录酶(RT)直接连接并通过使此酶的催化端断裂来阻断RNA依赖和DNA依赖的DNA聚合酶活性[1-2]. 奈韦拉平不与底物或三磷酸核苷产生竞争. 我们旨在摸索一种简便、快速、专属性高的测定奈韦拉平血浆浓度的方法.1材料和方法1.1材料安捷伦1100液相色谱仪(美国安捷伦公司生产);低温高速离心机(德国SIGMA公司生产);振荡器(美国科尔帕默仪器公司生产);万分之一天平(上海衡平仪器仪表厂生产). 甲醇、乙腈,色谱纯,Merck公司生产;磷酸、磷酸二氢钠、三氯乙酸、三乙胺,AR级,上海化学试剂公司生产. 受试制剂:国产奈韦拉平胶囊剂(商品名艾极),批号0309001,每粒200 mg,浙江华海药业股份有限公司生产;国产奈韦拉平片剂(商品名艾太),批号0402004,每片200 mg,浙江华海药业股份有限公司生产;参比制剂:进口奈韦拉平片(商品名维乐命),批号304185d,每片200 mg,德国勃林格殷格翰生产;奈韦拉平标准品,批号556503002,含量99.8%,浙江华海药业股份有限公司提供.1.2方法1.2.1色谱条件色谱柱: Hypersil C18柱(150 mm×4.6 mm, 0.5μm);流动相:乙腈: 0.01 mol/L磷酸二氢钠(含0.01 mol/L三乙胺,磷酸调pH至5.0)=18:82;流速: 1.0 mL/min;柱温:45℃;检测波长: 240 nm;进样量:25 μL;灵敏度: 0.01AUFS.1.2.2定性获取空白血浆、加入标准品的血浆样品和待测血浆样品的二维色谱图,比较所测成分的保留时间及检测波长是否一致.1.2.3血浆样品的预处理取0.2 mL血浆样品,加入20 μL 500 mL/L三氯乙酸,漩涡振荡,4℃下离心5 min(15000 r/min),取上清液25 μL进样,作HPLC分析.1.2.4标准曲线的制备取健康人的空白血浆0.2 mL数份,加入奈韦拉平标准品,使其浓度分别为0.1, 0.5, 1.0, 2.0, 3.5和5.0 mg/L,按血浆样品处理项处理后作HPLC分析. 以样品峰面积(A)对药物浓度(C)进行线性回归,计算回归方程.1.2.5回收率与精密度考察配制含奈韦拉平的标准品0.2, 2.5, 4.5mg/L的系列血样,日内各浓度重复进样5次,日间各浓度测定1次,连续测定5 d,以所测的浓度与已知浓度相比,计算血样中奈韦拉平的相对回收率和日内、日间精密度.1.2.6药代动力学参数计算Cmax, tmax用实测值表示,AUC0-t用梯形法计算. AUC(0-∞)=AUC(0-t)+Ct/λZ, Ct为最后一点的血药浓度,λZ为末端相消除速度常数,t1/2可按t1/2=0.693/λZ求出.2结果2.1色谱分离与定性从加入标准品的血浆样品的二维色谱图可知,奈韦拉平在12.165 min出峰,比较空白血浆、加入标准品的血浆样品和待测血浆样品的色谱图,奈韦拉平的保留时间基本一致,证明定性成功(图 1).A:空白血浆;B:加入标准品的血浆样品;C:待测血浆样品.图1人血浆中奈韦拉平的高效液相色谱图2.2线性关系回归方程: C=81.45A+58.65,相关系数(r)=0.9995(n=6),线性范围0.1~5.0 mg/L. 相关系数的显著性检验P<0.001,说明待测成分在所选的范围内线性关系良好. 本色谱条件下的最低检测浓度为0.1 mg/L.2.3专属性本方法专属性高,在该色谱条件下,测得奈韦拉平保留时间为12.165 min,药物峰形良好,血浆中内源性杂质不干扰奈韦拉平的测定.2.4精密度和回收率血样中奈韦拉平的相对回收率和日内、日间精密度,其结果见表1. 血样中奈韦拉平的相对回收率为99.41%~100.53%,日内、日间精密度小于10%,符合生物样品分析的有关要求.表1人血浆中奈韦拉平的精密度和回收率2.5血药浓度时间曲线采用二重3*3拉丁方设计,24名受试者按照随机顺序接受3种药物(奈韦拉平国产胶囊剂、片剂与进口片剂)200 mg后的平均血药浓度时间曲线见图2. 结果显示,血样中奈韦拉平实测浓度均在本试验线性范围内.图2奈韦拉平的平均血药浓度时间曲线3讨论测定人血浆中奈韦拉平浓度的方法有高效液相色谱法[3-4]和离子对色谱法[5]. 我们采用高效液相色谱法,血浆内源性杂质不干扰药物的测定. 采用磷酸盐缓冲溶液,调节pH=5,以获得较好的分离度和较短的保留时间. 该方法符合人体药代动力学研究生物样品测定的方法学要求.本实验中,对于流动相的选择和药物的萃取进行了大量探索工作,固相萃取可得到较高和较稳定的回收率[6],却代价昂贵,应用500 mL/L三氯乙酸作为萃取溶剂,回收率高而且稳定,以乙腈和磷酸盐溶液作为流动相[7],药物峰受到血浆内源性杂质峰干扰,磷酸盐溶液中加入三乙胺,消除杂质峰的干扰,实验方法有较高专属性.奈韦拉平是一种抗艾滋病新药,有较多不良反应,会引起肝毒性和耐受性[8-9]. 本次试验过程因志愿者出现不良反应而脱落4例.【参考文献】[1] Tebas P, Yarasheski K, Henry K, et al. Evaluation of the virological and metabolic effects of switching protease inhibitor combination antiretroviral therapy to nevirapinebased therapy for the treatment of HIV infection[J]. AIDS Res Hum Retroviruses, 2004,20(6):589-594.[2] Manosuthi W, Sungkanuparph S, Vibhagool A, et al. Nevirapineversus efavirenzbased highly active antiretroviral therapy regimens in antiretroviralnaive patients with advanced HIV infection[J]. HIV Med, 2004,5(2):105-109.[3] Laurito TL, Santagada V, Caliendo G, et al. Nevirapine quantification in human plasma by highperformance liquid chromatography coupled to electrospray tandem mass spectrometry. Application to bioequivalence study [J]. J Mass Spectrom, 2002,37(4):434-441.[4] Lopez RM, Pou L, Gomez MR, et al. Simple and rapid determination of nevirapine in human serum by reversedphase highperformance liquid chromatography[J]. J Chromatogr B Biomed Sci Appl, 2001,751(2):371-376.[5] Fan B, Stewart JT. Determination ofzidovudine/lamivudine/nevirapine in human plasma using ionpair HPLC [J]. J Pharm Biomed Anal, 2002,28(5):903-908.[6] Colombo S, Beguin A, Marzolini C, et al. Determination of the novel nonpeptidic HIVprotease inhibitor tipranavir by HPLCUV after solidphase extraction[J]. J Chromatogr B Analyt Technol Biomed Life Sci, 2006,832(1):138-143.[7] Notari S, Bocedi A, Ippolito G, et al. Simultaneous determination of 16 antiHIV drugs in human plasma by highperformance liquid chromatography[J]. J Chromatogr B Analyt Technol Biomed Life Sci, 2006,831(12):258-266.[8] Gokengin D, Yamazhan T. Hepatic adverse events during highly active antiretroviral therapy containing nevirapine: A casereport [J]. Ann Clin Microbiol Antimicrob, 2002,1:1.[9] Manosuthi W, Chumpathat N, Chaovavanich A, et al. Safety and tolerability of nevirapinebased antiretroviral therapy in HIVinfected patients receiving fluconazole for cryptococcal prophylaxis: A retrospective cohort study [J]. BMC Infect Dis, 2005,5:67.。
UPLC法测定注射用特利加压素中有关物质的含量

UPLC法测定注射用特利加压素中有关物质的含量Content Determination of Related Substances in Terlipressin for Injection by UPLCXUE Qiaoru,YUAN Jinye,DING Liuyang,ZHENG Weijun,DENG Feng(Guangdong Institute for Drug Control/NMPA Key Laboratory of Quality Control of Blood Products/Key Laboratory of Quality Control and Research of Blood Products,Guangzhou,*****,China)*****T *****VE:To establish UPLC method for the content determination of related substances in Terlipressin for injection. *****:UPLC method was used to determine the contents of related substances in 5 batches of Terlipressin for injection. The separation was performed on Xtimate UPLC C18 column with mobile phase A consisted of ammonium sulfate buffer (pH 2.3)-methanol (90 ∶ 10,V/V)and mobile phase B consisted of ammonium sulfate buffer (pH 2.3)-methanol (60 ∶ 40,V/V)(gradient elution)at the flow rate of 0.2 mL/min. The detection wavelength was set at 210 nm,and sample size was 5 μL. *****:The linear range of impurity A,B,C,D,F,H,I,K,L and N were 0.43-3.86,0.44-3.95,0.44-3.97,0.45-4.08,0.45-4.05,0.50-4.50,0.47-4.26,0.47-4.23,0.46-4.13,0.44-3.96 μg/mL (r≥0.999 7),respectively. The detection limits were 0.04,0.04,0.05,0.04,0.05,0.05,0.05,0.05,0.04 μg/mL. The quantitation limits were 0.13,0.13,0.14,0.13,0.15,0.14,0.14,0.14,0.13 μg/mL,respectively. RSDs of precision,reproducibility and stability tests were all lower than 8%. The average recoveries were 94.95%,97.81%,101.88%,95.26%,93.40%,102.48%,104.26%,102.31%,96.42%,90.42%,with RSDsof 1.89%,1.86%,0.68%,1.30%,1.98%,3.36%,1.26%,1.30%,1.19%,1.40% (n=9),respectively. Total contents of impurities in 5 batches of Terlipressin for injection were all lower than 4%. *****IONS:Established method is rapid,simple,accurate and specific,which can be used for the quantitative analysis for related substances in Terlipressin for injection.*****S UPLC; Terlipressin for injection; Related substance; Content determination注射用特利加压素的主成分为醋酸特利加压素(Terlipressin),其化学名为L-甘氨酰-L-甘氨酰-L-甘氨酰-L-半胱氨酰-L-酪氨酰-L-苯丙氨酰-L-谷氨酰氨酰-L-天冬酰氨酰-L-半胱氨酰-L-脯氨酰-L-赖氨酰-L-甘氨酰胺醋酸盐(4→9-二硫环)。
HPLC法测定奈韦拉平的血药浓度

HPLC法测定奈韦拉平的血药浓度
周细根;陈汇
【期刊名称】《实用临床医学》
【年(卷),期】2006(007)009
【摘要】目的:建立高效液相色谱法测定奈韦拉平血药浓度的方法.方法:采用Hypersil C18柱(150 mm× 4.6 mm,0.5μm);流动相;乙腈:0.01 mol/L磷酸二氢钠(含0.01 mol/L三乙胺,磷酸调pH至5.0)=18:82;流量:1.0mL/min;检测波
长:240nm.结果:线性范围0.1~5.0 mg/L(r=0.9996);最低检测浓度为0.1 mg/L;日内、日间相对标准差(RSD%)均小于10%;低、中、高三个浓度的提取回收率分别为99.98%、99.41%和100.53%.结论:该法简便、快速、准确,专属性高,适用于奈韦拉平药动学和生物等效性研究.
【总页数】3页(P16-17,19)
【作者】周细根;陈汇
【作者单位】井冈山学院医学院,江西,吉安,343000;华中科技大学同济医学院临床药理系,湖北,武汉,430030
【正文语种】中文
【中图分类】R917.1
【相关文献】
1.HPLC法测定奈韦拉平片的含量及有关物质 [J], 赖红宁;冯玉欢;梁松庆
2.高效液相色谱法测定奈韦拉平的血药浓度 [J], 梁生林;周细根;温永顺
3.液液萃取-HPLC法和在线萃取-HPLC法测定卡马西平血药浓度的比较研究 [J], 邓利娟;王峰;谢娇
4.UHPLC-MS/MS法测定氟苯尼考明胶纳米药物在小鼠体内的血药浓度 [J], 亓馨怡;侯冰玉;李建科;姚霄月;黄晶晶;李臣贵
5.用HPLC法测定奈韦拉平含量的不确定度评定 [J], 张平;余杰
因版权原因,仅展示原文概要,查看原文内容请购买。
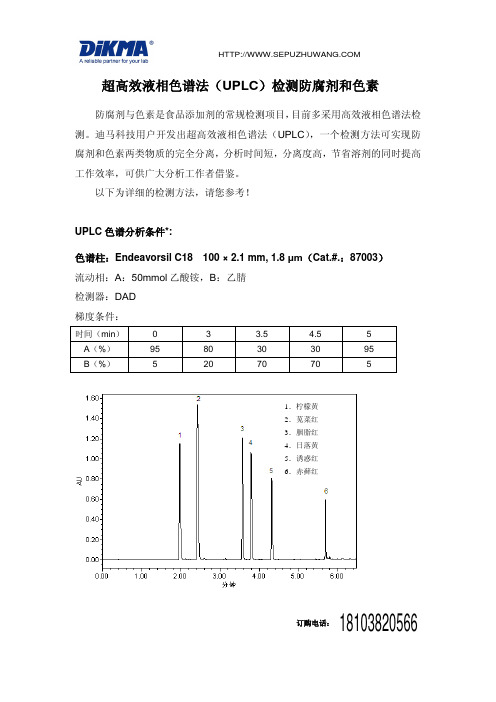
超高效液相色谱法(uplc)检测防腐剂和色素
乙腈 HPLC 级
2 上海:021-6126 3966 天津:400-633-6606 南京:025-8347 9007
4L
广州:020-8559 3520 青岛:0532-8372 5230 石家庄:0311-6668 6220
50138
乙酸铵 HPLC 级 通用色谱产品 瓶架/蓝色 瓶架/白色 样品瓶(棕色/螺纹) 样品瓶盖/含垫(已经组装) HPLC 进样针
广州:020-8559 3520 青岛:0532-8372 5230 石家庄:0311-6668C)检测防腐剂和色素
防腐剂与色素是食品添加剂的常规检测项目, 目前多采用高效液相色谱法检 测。迪马科技用户开发出超高效液相色谱法(UPLC) ,一个检测方法可实现防 腐剂和色素两类物质的完全分离,分析时间短,分离度高,节省溶剂的同时提高 工作效率,可供广大分析工作者借鉴。 以下为详细的检测方法,请您参考!
100 g 50 孔 50 孔 2 mL, 100/pk 100/pk 25 μL
52401B 52401A 5323 5325 H80465
订购电话:
北京:400-608-7719 沈阳:024-2294 3513 成都:028-8661 2625
3 上海:021-6126 3966 天津:400-633-6606 南京:025-8347 9007
订购电话: 北京:400-608-7719 沈阳:024-2294 3513 成都:028-8661 2625
规格 13 mm,0.22 μm 100/pk 13 mm,0.45 μm 100/pk
100 × 2.1 mm, 1.8 μm
25mg ≥99.0% (HPLC) 25mg ≥98.0% (HPLC) 25mg ≥99.0% (HPLC) 25mg ≥95% (HPLC) 25mg ≥98.0% (HPLC) 25mg ≥98.0% (HPLC) 1g 1g 1g
用HPLC法测定奈韦拉平含量的不确定度评定

用HPLC法测定奈韦拉平含量的不确定度评定
张平;余杰
【期刊名称】《药学服务与研究》
【年(卷),期】2007(7)6
【摘要】目的:对HPLC法测定奈韦拉平含量的测量不确定度进行分析,以期找出影响不确定度的因素,为评价检测报告提供科学依据。
方法:用HPLC法测定原料药奈韦拉平的含量,并根据《(JJF1059-1999)测量不确定度评定与表示》中有关规定评估其不确定度。
结果:本次实验的不确定度评估为1.2%。
结论:本次实验的不确定度主要由供试品溶液峰面积的重复性测量引起的。
【总页数】3页(P436-438)
【关键词】奈韦拉平;色谱法,高压液相;测量不确定度
【作者】张平;余杰
【作者单位】上海市崇明县食品药品检验所;上海三维生物技术有限公司
【正文语种】中文
【中图分类】R927.2
【相关文献】
1.HPLC法测定赖诺普利片含量及含量均匀度的不确定度评定 [J], 谢琼玉
2.HPLC-MS法测定药品含量的不确定度评定 [J], 金绍杰;杜昌冬;宋玉琳;叶晖;张跃锋
3.HPLC法测定盐酸利多卡因注射液含量的不确定度评定 [J], 张志超;袁晓转;韩慧
丽
4.HPLC法测定酒石酸美托洛尔片含量的测量不确定度评定 [J], 蔡宣越
5.HPLC-示差折光检测法测定盐酸克林霉素棕榈酸酯分散片含量的不确定度评定[J], 庞赛;刘晶晶;李玉兰;刘敏;王铁杰
因版权原因,仅展示原文概要,查看原文内容请购买。
奈韦拉平简介

奈韦拉平简介目录•1拼音•2英文参考•3奈韦拉平药典标准o 3.1品名▪ 3.1.1中文名▪ 3.1.2汉语拼音▪ 3.1.3英文名o 3.2结构式o 3.3分子式与分子量o 3.4来源(名称)、含量(效价)o 3.5性状o 3.6鉴别o 3.7检查▪ 3.7.1有关物质▪ 3.7.2残留溶剂▪ 3.7.3水分▪ 3.7.4炽灼残渣▪ 3.7.5重金属o 3.8含量测定▪ 3.8.1色谱条件与系统适用性试验▪ 3.8.2测定法o 3.9类别o 3.10贮藏o 3.11制剂o 3.12附杂质o 3.13版本•4参考资料1拼音nài wéi lā píng2英文参考Nevirapine[湘雅医学专业词典]3奈韦拉平药典标准3.1品名3.1.1中文名奈韦拉平3.1.2汉语拼音Naiweilaping3.1.3英文名Nevirapine3.2结构式3.3分子式与分子量C15H14N4O 266.303.4来源(名称)、含量(效价)本品为11环丙基5,11二氢4甲基6H二吡啶并[3,2,b:2',3'e] [1,4]二氮杂䓬6酮。
按无水物计算[1],含C15H14N4O应为98.0%~102.0%。
3.5性状本品为白色或类白色结晶性粉末;无臭。
本品在乙醇或甲醇中微溶[1],在水中几乎不溶。
3.6鉴别(1)在含量测定项下记录的色谱图中,供试品溶液主峰的保留时间应与对照品溶液主峰的保留时间一致。
(2)本品的红外光吸收图谱应与对照的图谱(《药品红外光谱集》1159图)一致。
3.7检查3.7.1有关物质取含量测定项下的供试品贮备液,作为供试品溶液;精密量取供试品溶液1ml,置100ml量瓶中,用流动相稀释至刻度,摇匀,再精密量取5ml,置50ml量瓶中,用流动相稀释至刻度,摇匀,作为对照溶液。
照含量测定项下的色谱条件,取对照溶液50μl注入液相色谱仪,调节检测灵敏度,使主成分色谱峰的峰高约为满量程的25%。
UPLC法测定奈韦拉平的含量及有关物质

缩短为 13 min。经方法学考察和样品测定,结果表 明所建方法可准确测定奈韦拉平原料药的含量及有 关物质。
有关奈韦拉平的合成和制备方式国内外均有报 道,1991 年 Hargrave 等最早报道了奈韦拉平的合成 路线[7]。1995 年 Schneider 等 改 进 了 以 上 合 成 路 线[8],主要体现在第 2 步化合物 3 与环丙胺反应时 添加了中和剂( 碱土金属、锌氧化物或氢氧化物,如 氧化钙) ,以防止反应中生成的氯化氢与环丙胺成 盐,提高反应产率,并使用了无毒的高沸点溶剂二甘 醇二甲醚( DMDE) ,该法由于收率高,污染少,已成 为国内外奈韦拉平工业化生产的主要方法。此后, 关于奈韦拉平的工业化制备合成工艺的改进均以此 方法为基础进行调整,主要涉及中间过程的步骤以 及反应物质和催化剂·mL -1 的范围内线性关系良好( r = 0. 999,n = 6) ,平均回收率为 99. 6% ,RSD = 2. 0% ( n = 9) 。结论: 本方法快
速、简便、准确,专属性强。
关键词: 超高效液相色谱法; 奈韦拉平; 含量测定; 有关物质
中图分类号: R921. 2
用甲醇溶解并定量稀释成系列浓度的溶液。依法注 入液相色谱仪,记录色谱图,绘制标准曲线。结果表 明奈韦拉平在 0. 16 ~ 0. 96 mg·mL - 1 的范围内与峰 面 积 呈 良 好 的 线 性 关 系,回 归 方 程 为 Y = 4 721 447. 5X + 76 760. 8( r = 0. 999) ; 杂质 A 在 0. 04 ~ 4. 0 μg·mL - 1 范围内与峰面积呈良好的线 性关 系,回 归 方 程 为 Y = 4 953. 5X + 2. 5 ( r = 1. 000) ; 杂质 B 在 0. 04 ~ 4. 0 μg·mL -1 范围内与峰 面积呈良好的线性关系,回归方程为 Y = 4 550. 1X + 6. 1( r = 0. 999) ; 杂质 C 在 0. 04 ~ 4. 0 μg·mL -1 范围 内与峰 面 积 呈 良 好 的 线 性 关 系,回 归 方 程 为 Y = 4 550. 1X + 6. 1( r = 1. 000) 。 2. 7 进样精密度试验
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
中国药品标准 2015 年第 16 卷第 4 期 270
1. 酸破坏; 2. 碱 破 坏; 3. 高 温 破 坏; 4. 氧 化 破 坏; 5. 光 照 破 坏; 6. 未经破坏
图 3 破坏试验色谱图( 批号 C5021-12-045)
2. 6 线性关系考察 精密称取奈韦拉平、杂质 A,B,C 对照品适量,
文献标识码: A
文章编号: 1009 - 3656( 2015) - 4 - 268 - 3
Determination of Nevirapine and Related Substances by UPLC
Xue Qiaoru,Liang Weiyang* ,Luo Zhuoya( Guangdong Institute for Food and Drug Ccontrol,Guangzhou 510180)
校正 因子
1. 3 1. 0 1. 0 0. 9
2. 4 专属性试验 2. 4. 1 酸破坏 取供试品 20 mg,加 1 mol·L -1 盐酸 溶液 10 mL,室温放置 3 h,加 1 mol·L -1 氢氧化钠溶 液 10 mL 中和,加流动相至 50 mL,作为供试品溶液。 2. 4. 2 碱破坏 取供试品 20 mg,加 1 mol·L - 1 氢 氧化钠溶液 10 mL,室温放置 3 h,加 1 mol·L - 1 盐 酸溶液 10 mL 中和,加流动相至 50 mL,作为供试品 溶液。 2. 4. 3 高温破坏 取供试品 20 mg,加流动相 20 mL,水浴加热 3 h,加流动相至 50 mL,作为供试品 溶液。 2. 4. 4 氧化破坏 取供试品 20 mg,加流动相 20 mL,加浓过氧化氢溶液 1 mL,室温放置 3 h,加流动 相至 50 mL,作为供试品溶液。 2. 4. 5 光照破坏 取供试品 20 mg,加流动相 50 mL,置日光灯下光照 24 h,作为供试品溶液。
Drug Standards of China 2015,Vol. 16 No. 4
中国药品标准 2015 年第 16 卷第 4 期 269
图 1 奈韦拉平主要杂质结构式
1 仪器与试药 Waters ACQUITY 超高效液相色谱系统。 奈韦拉平对照品( 中国药品生物制品检定所,
批号 100641-200401,含 量 100% ) ; 杂 质 A 对 照 品 ( USP H1K372,含 量 100% ) ; 杂 质 B 对 照 品 ( USP H0J192,含量 100% ) ; 杂质 C 对照品( EP 2957,含量 99. 4% ) ; 奈韦拉平原料药( 企业 A,批号 C5021-12045,C5021-12-046,C5021-12-047; 企 业 B,批 号 120858,120859,120860; 企 业 C,批 号 1011001, 1009003,1006004; 企业 D,批号 1202013,1208079, 1208080) ; 乙腈为 Honeywell N70A1H 色谱纯,磷酸 二氢铵为分析纯,水为 millipore 超纯水。
图 2 系统适用性试验色谱图
表 1 系统适用性试验结果
成分
杂质 B 奈韦拉平 杂质 A 杂质 C
保留时间 相对保留
/ min
时间
0. 796 1. 170 1. 945 3. 821
0. 68 1. 00 1. 66 3. 27
理论 板数
7 490 8 037 8 954 9 599
分离 度
/ 8. 6 11. 8 15. 9
奈韦拉平现行标准为 WS1 -( X-064 ) -2005Z,未 对已知杂质进行控制; 国外标准《美国药典》35 版[3] ( USP 35) 、《英国药典》2012 年版[4]( BP 2012) 、《欧 洲药典》7. 0 版[5]( EP 7. 0) 、《国际 药 典》4. 0 版[6] ( IP 4. 0) 均有收载,且采用 HPLC 法对已知杂质 A, B,均进行了控制,分析时间规定约为 80 min。为 缩短测试时间,减少试剂消耗,本实验采用超高效液 相系统( UPLC) 对现行标准进行了优化,分析时间
奈韦拉平( nevirapine) 是一种新型人体免疫缺 陷病毒( HIV-1) 的非核甘类逆转录酶抑制剂,奈韦 拉平与 HIV-1 的逆转录酶直接连接并且通过使此酶 的催化端破裂来阻断 RNA 依赖和 DNA 依赖的 DNA 聚合酶活性,从而抑制逆转录酶活性,减少病毒复制, 降低病毒对机体功能的破坏。该药品由德国 Boehringer Ingelheim 公 司 研 发,于 1994 年 获 得 美 国 专 利[1],1996 年 9 月美国 FDA 批准 上 市,商 品 名 Viramune,主要 用 于 艾 滋 病 的 预 防 和 治 疗[2]。 该 药 品 2001 年进入我国,目前国内也有药品生产企业生产。
2 方法与结果 2. 1 色谱条件
色谱柱为 Acquity UPLC TSS T3 C18 ( 2. 1 mm × 50 mm,1. 8 μm) ; 流动相为 10 mmol·L - 1 磷酸二氢 铵溶液( 取磷酸二氢铵 1. 15 g,加水 800 mL 使溶解, 用 1 mol·L - 1 氢氧化钠溶液调节 pH 值至 5. 0,再加 水稀释至 1 000 mL) -乙腈( 80 ∶ 20) ; 柱温 35 ℃ ; 检 测波长 220 nm; 进样量 5 μL。 2. 2 溶液的制备 2. 2. 1 供试品溶液 精密称取供试品适量,加甲醇 溶解并制成每 1 mL 中含 0. 4 mg 的溶液,即得( 必要 时可超声溶解) 。 2. 2. 2 对照溶液 精密量取供试品溶液 1 mL,置 100 mL 量瓶中,加甲醇稀释至刻度,摇匀,精密量取 1 mL,置 10 mL 量瓶中,加甲醇稀释至刻度,摇匀, 即得。 2. 3 系统适用性试验
破坏性试验的结果表明奈韦拉平具有较好的稳 定性,见图 3。上述供试品溶液测定结果与未经破 坏的供试品溶液的主峰面积基本相当,表明在该条 件下,奈韦拉平相对稳定,且该条件下奈韦拉平与相 邻峰均有较好的分离,满足测定要求。 2. 5 检测限与定量限
在上述色谱条件下,取“2. 3”项下系统适用性 试验溶液逐步稀释,按信噪比 S / N≈3 测定检出限, 按信噪比 S / N≈10 测定定量限。结果杂质 B,奈韦 拉平、杂质 A 的检出限为 0. 01 μg·mL - 1 ,定量限为 0. 03 μg·mL - 1 ; 杂质 C 的检出限和定量限分别为 0. 012 μg·mL - 1 和 0. 036 μg·mL - 1 。
缩短为 13 min。经方法学考察和样品测定,结果表 明所建方法可准确测定奈韦拉平原料药的含量及有 关物质。
有关奈韦拉平的合成和制备方式国内外均有报 道,1991 年 Hargrave 等最早报道了奈韦拉平的合成 路线[7]。1995 年 Schneider 等 改 进 了 以 上 合 成 路 线[8],主要体现在第 2 步化合物 3 与环丙胺反应时 添加了中和剂( 碱土金属、锌氧化物或氢氧化物,如 氧化钙) ,以防止反应中生成的氯化氢与环丙胺成 盐,提高反应产率,并使用了无毒的高沸点溶剂二甘 醇二甲醚( DMDE) ,该法由于收率高,污染少,已成 为国内外奈韦拉平工业化生产的主要方法。此后, 关于奈韦拉平的工业化制备合成工艺的改进均以此 方法为基础进行调整,主要涉及中间过程的步骤以 及反应物质和催化剂的改进等。
中国药品标准 2015 年第 16 卷第 4 期 268
·方法学研究·
Drug Standards of China 2015,Vol. 16 No. 4
UPLC 法测定奈韦拉平的含量及有关物质
薛巧如,梁蔚阳* ,罗卓雅( 广东省食品药品检验所,广州 510180)
摘要 目的: 改进奈韦拉平的含量和有关物质测定方法。方法: 采用 Acquity UPLC TSS T3 C18 ( 2. 1 mm × 50 mm,1. 8 μm) ,流 动相为 10 mmol·L - 1 磷酸二氢铵溶液( pH 5. 0) -乙腈( 80∶ 20) ,流量为 0. 7 mL·min - 1 ,检测波长为 220 nm。结果: 奈韦拉平在
杂质 A,B,C 均为原料合成过程中产生的主要 工艺杂质,结构式见图 1。国内外产品中均含有上 述三种已知杂质。
作者简介: 薛巧如,副主管药师; 研究方向: 生化药品的检验与质量控制。 作者简介: 梁蔚阳,主任药师; Tel: 020 - 81848209; E-mail: wl-1023@ 163. com
0. 16 ~ 0. 96 mg·mL -1 的范围内线性关系良好( r = 0. 999,n = 6) ,平均回收率为 99. 6% ,RSD = 2. 0% ( n = 9) 。结论: 本方法快
速、简便、准确,专属性强。