产品稳定性试验方案
稳定性考察方案
文件名称稳定性考察方案文件编码颁发部门质量保证部版本01 执行日期起草:日期:审核:日期:批准:日期:分发部门质量保证部质量控制中心生产管理部设备动力部供应部市场部综合部财务部生产车间份数文件内容1.目的2.责任人3.适用范围4. 内容1.目的建立稳定性考察方案,对产品品种、批号、有效期进行考察,为产品制定有效期提供依据。
2.责任人留样观察管理员3.适用范围已经生产的全部产品。
4. 内容1.稳定性考察方法:长期试验长期试验是在接近药品实际贮存条件下进行,其目的是为制订药品的有效期提供依据。
供试品要求3批,市售包装,在温度25℃±4℃,相对湿度60%±15%的条件下放置36个月,分别于第0月、3月、6月、9月、12月、24月、36月,按稳定性考察检查项目进行检查,将结果与0月比较观察其质量否有显著性差异。
2检测时间的规定2.1取样检查时间:6个月允许±15天;12个月后允许±30天。
2.2样品取出先放在常温下,一般要求一周内完成检测;温湿度敏感的药品要及时检测。
3.稳定性试验方案的实施3.1在药品稳定性试验前,留样管理员应检查其储存条件是否合格。
检查合格后将样品存入指定位置。
3.2留样管理员依据稳定性试验方案确定的考察品种、考察月份、检查项目,及时取样、申请检验。
3.3稳定性考察检查项目:考察月份检查项目0 全检3 全检6 全检9 全检12 全检24 全检36 全检其中,性状,主要检查中药材或饮片外观颜色是否符合要求,是否有生虫、发霉迹象。
3.4 QC收到请检样品后,依据稳定性考察检查项目,及时安排检测。
检测结束后,根据检测结果,出具检验报告书给留样观察员。
3.5稳定性试验结果出现异常情况,调查原因,并将情况及时向质量管理负责人汇报。
如果只有一项次要的检测指标有变化(如颜色、水分变化,但仍在标准之内),则继续进行稳定性试验,观察其变化趋势。
若发生显著变化,则应调整试验条件,重新进行试验。
产品稳定性试验加速试验方案

文件制修订记录鉴于目前国家尚无出台食品添加剂稳定性试验指导原则,参照《中国药典》2020年版9001原料药物与制剂稳定性试验指导原则,根据公司产品质量稳定性考察计划,结合XXX产品特点和公司总部要求,现拟定XXX稳定性试验加速试验方案。
1、目的:在加速条件下进行加速试验,目的是通过加速产品的化学或物理变化,考察市售包装品在有效期内的质量稳定性,为产品设计、包装、运输、贮存提供必要的资料。
加速试验所用包装应采用模拟小包装,但所用材料与封装条件应与大包装一致。
2、范围:本方案适用于公司生产的市售包装XXX产品。
3、责任:质量部经理负责稳定性考察加速试验方案与报告的起草和批准,QC 人员按照本方案完成实验并报告检验结果。
4、质量标准依据与考察项目:5.1考察批次及样品数:本产品市售包装成品规格为25千克/铝箔袋,选择连续生产的三批模拟市售包装小袋(50g/铝箔袋)产品,每批次10袋,三批次共30袋。
5.2考察方法5.2.1贮存地点:XXX公司稳定性试验室。
5.2.2试验条件:在温度40℃±2℃、相对湿度75%±5%的条件下放置6个月。
在试验期间第1个月、2个月、3个月、6个月末分别取样一次,按稳定性考察项目检测。
在上述条件下,如6个月内供试品经检测不符合制订的质量标准,则应在中间条件下即在温度30℃±2℃、相对湿度65%±5%的情况下(可用Na2Cr04饱和溶液,30℃,相对湿度64.8%)进行加速试验,时间仍为6个月。
样品留样管理人员应在检测日的当天将样品取出交给检验人员,检验人员应在样品接收后立即安排进行测定,并将检验结果报告QC主管。
5.2.4合格标准5.2.4.1按照该稳定性考察方案完成所有测试,测试结果合格;5.2.4.2没有偏差,或者偏差的调查结果和产品质量无关。
6、产品稳定性考察报告6.1试验报告内容的要求应包括产品考察指标与合格标准、考察过程中出现的偏差及处理情况、数据统计、考察结论、附表(实验数据)。
产品稳定性试验方法
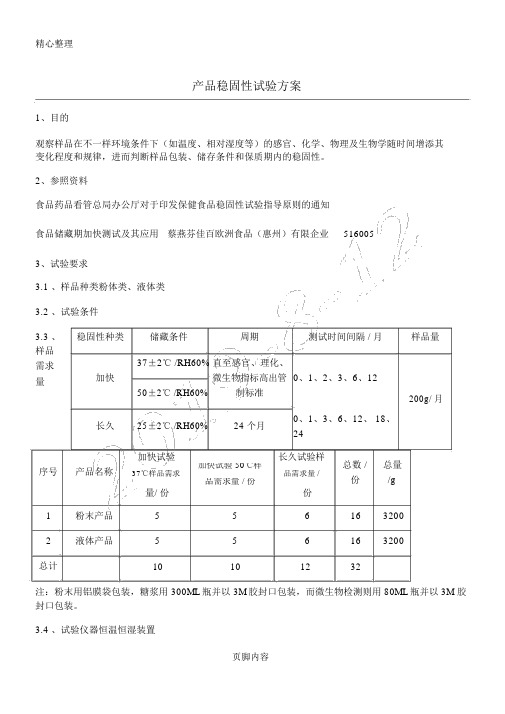
产品稳固性试验方案1、目的观察样品在不一样环境条件下(如温度、相对湿度等)的感官、化学、物理及生物学随时间增添其变化程度和规律,进而判断样品包装、储存条件和保质期内的稳固性。
2、参照资料食品药品看管总局办公厅对于印发保健食品稳固性试验指导原则的通知 食品储藏期加快测试及其应用 蔡燕芬佳百欧洲食品(惠州)有限企业 5160053、试验要求3.1 、样品种类粉体类、液体类3.2 、试验条件3.3 、 稳固性种类储藏条件 周期测试时间间隔 / 月样品量样品 37±2℃ /RH60% 直至感官、理化、需求加快微生物指标高出管 0、1、2、3、6、12量50±2℃ /RH60%制标准200g/ 月长久25±2℃ /RH60%24 个月0、1、3、6、12、 18、 24加快试验长久试验样总数 /总量序号产品名称加快试验 50℃样37℃样品需求品需求量 /份/g 品需求量 / 份份量/ 份1 粉末产品 5 5 6 16 32002 液体产品5 56 16 3200总计10101232注:粉末用铝膜袋包装,糖浆用 300ML 瓶并以 3M 胶封口包装,而微生物检测则用 80ML 瓶并以 3M 胶封口包装。
3.4 、试验仪器恒温恒湿装置页脚内容4、实验方案方案恒温恒湿箱加快试验长久试验同一批次的试验产品,温度30把留样室调整到试验所需的温方案一 1 个工作室度与50度两种不一样的环境轮番湿度,样品寄存于留样室进行做试验试验同一批次的试验产品,温度30把留样室调整到试验所需的温方案二 2 个工作室度与50度两种不一样的环境分别湿度,样品寄存于留样室进行在不一样的工作室同时进行试验试验一个工作室将设为温度30 度的环境做试验方案三 3 个工作室另一个工作室用作长久试验一个工作室将设为温度50 度的环境做试验注: 1、仪器尺寸的选择,因为实验室空间有限,在选购的恒温恒湿箱空间尺寸时有所限制,而我们企业的产品种类许多,试验时只好分批次来进行。
稳定性试验计划

稳定性试验计划
1. 目的
稳定性试验旨在评估样品在特定环境条件下的质量变化情况,从而预测产品在建议的储存条件下的有效期。
2. 试验样品
- 样品描述(如批号、生产日期等)
- 样品数量
- 包装材料和容器
3. 储存条件
- 长期储存条件(25±2°/60±5%)
- 中期储存条件(30±2°/65±5%)
- 加速储存条件(40±2°/75±5%)
4. 试验时间点
- 初始时间点
- 3个月
- 6个月
- 9个月
- 12个月
- 18个月
- 24个月
- (根据具体情况设置时间点)
5. 评价指标
- 物理性状(颜色、气味、状态等)
- 化学鉴别
- 含量测定
- 其他指标(如需要)
6. 判据
按照相关法规和指导原则,制定合理的判据标准。
7. 样品处理
规定不同时间点取样、检测流程。
8. 报告
及时整理数据,按期编写稳定性试验报告。
以上为一般的稳定性试验计划框架,具体内容需根据产品特性和相关法规要求进行调整。
定期开展稳定性试验对于确保产品质量、评估有效期至关重要。
稳定性试验方案

原料药与药物制剂稳定性试验指导原则稳定性试验的目的是考察中药在温度、湿度、光线、微生物的影响下随时间变化的规律。
为中药的生产、包装、贮存、运输条件提供科学依据,同时根据试验结果建立药品的有效期。
稳定性试验的基本要求有以下几个方面:(1)稳定性试验包括加速试验与长期试验。
加速试验与长期试验要求用三批供试品进行。
(2)中药制剂的供试品应是放大试验的产品,其处方与工艺应与大生产一致。
每批放大试验的规模,丸剂应在10000g或10000丸左右、片剂10000片左右、胶囊剂10000粒左右,大体积包装的制剂(如静脉输液、口服液等)每批放大规模的数量至少应为各项试验所需总量的10倍。
特殊品种、特殊剂型所需数量,根据情况,灵活掌握。
(3)供试品的质量标准应与各项基础研究及临床验证所使用的供试品质量标准一致。
(4)加速试验与长期试验所用供试品的容器和包装材料及包装方式应与上市产品一致。
(5)研究中药稳定性,要采用专属性强、准确、精密、灵敏的分析方法并对方法进行验证,以保证中药稳定性试验结果的可靠性。
在稳定性试验中,应重视降解产物的检查。
(6)由于放大试验比大规模生产的数量要小,故申报者应在获得批准后,从放大试验转入大规模生产时,对最初通过生产验证的三批大规模生产的产品仍需进行加速试验与长期稳定性试验。
1、加速试验此项试验是在超常的条件下进行,其目的是通过加速中药的化学或物理变化,探讨中药的稳定性,为中药审评、工艺改进、包装、运输及贮存提供必要的资料。
供试品要求3批,按市售包装,在温度40℃±2℃,相对湿度75%±5%的条件下放置6个月。
所用设备应能控制温度±2℃,相对湿度±5%,并能对真实温度与湿度进行监测。
在试验期间第1个月、2个月、3个月、6个月末各取样一次,按稳定性重点考察项目检测。
在上述条件下,如6个月内供试品经检测不符合制订的质量标准,则应在中间条件下即温度30℃±2℃,相对湿度60%±5%的情况下进行加速试验,时间仍为6个月。
XXXX年度产品持续稳定性考察计划

质量管理部年度产品持续稳定性考察计划为考察我公司所有生产品种的质量稳定性,决定对所有生产品种进行长期稳定性考察试验,为确保患者能够安全、有效的用药和对各品种有效期确定等提供有效的数据。
一、稳定性考察要点药品剂型检验质量标准稳定性考察项目合格标准片剂《中国药典》2010年版一部、部颁标准性状、鉴别、重量差异、崩解时限、含量测定等符合各品种法定的质量标准颗粒剂《中国药典》2010年版一部性状、鉴别、水分(干燥失重)、溶化性、粒度、含量测定等胶囊剂部颁标准性状、鉴别、崩解时限、水分、含量测定等糖浆剂《中国药典》2010年版一部、部颁标准性状、鉴别、相对密度、PH值、含量测定酊剂《中国药典》2010年版一部性状、鉴别、相对密度、乙醇量、含量测定等二、具体方案1.对各品种生产的不同包装规格成品的正式开始生产前三批进行稳定性考察,以后每年对同一包装规格取一批次产品进行稳定性考察。
2.考察方法2.1、按市售包装,在药品标示的贮存条件下保存12个月(温度25℃±2℃、相对湿度60%±10%)。
每3个月取样一次,分别于0个月,3个月,6个月,9个月,12个月取样,按各剂型品种具体的稳定性考察项目进行检测。
12个月后,仍需继续考察,分别于18个月,24个月,36个月取样进行检测。
将结果与0个月比较以确定药品有效期。
2.2、由于实验数据的分散性,一般应按95%可信限进行统计分析,得出合理的有效期。
如三批统计数据分析结果差别较小,则取平均值为有效期。
若差别较大,则取其最短的为有效期。
同时能够明确各剂型品种的安全性、稳定性及有效性。
2.3、对于每种规格、每种内包装形式的药品,每年考察一个批次,若是原计划考察当月的指定包装规格的品种未生产,则选取离原计划最近月份所生产的相同内包装形式的品种进行稳定性考察,除非当年没有生产。
2.4、某些情况下,持续稳定性考察中应当额外增加批次数,如重大变更或生产和包装有重大偏差的药品应列入稳定性考察。
药物稳定性试验方案
***药物稳定性试验方案一、试验目的***药物稳定性试验的目的是考察***药物在温度、湿度、光线的影响下随时间变化的规律,考察其在影响因素试验、加速试验的条件下各项指标是否符合***药物产品质量标准的要求。
二、试验要求***药物稳定性试验的基本要求有以下几个方面:1、稳定性试验项目包括影响因素试验与加速试验。
2、***药物供试品可以从橡胶膏生产车间生产的同一批制剂中抽取,抽样数量符合稳定性试验要求。
3、加速试验与长期试验***药物所用供试品的容器和包装材料及包装方式应与上市产品一致。
4、***药物稳定性试验,要采用专属性强、准确、精密、灵敏的药物分析方法,以保证药物稳定性结果的可靠性。
三、试验方法(一)影响因素试验此项试验是在比加速试验更激烈的条件下进行。
其目的是考察***药物的固有稳定性,了解其在高温、高湿及低温条件下各项质量指标的稳定性及变化情况。
1.高温试验***药物置药物稳定性检查仪中,60℃温度下放置10天,于第5天和第10天取样,按***药物成品质量标准进行全检。
若***药物供试品有明显变化(如含量下降5%),则在40℃条件下同法进行试验。
若60℃无明显变化,不再进行40℃试验。
2.高湿度试验***药物置恒湿密闭容器中,在25℃分别于相对湿度90%±5%条件下放置10天,于第5天和第10天取样,按***药物成品质量标准进行全检,同时准确称量试验前后供试品的重量,以考察供试品的吸湿潮解性能。
若吸湿增重5%以上,则在相对湿度75%±5%条件下,同法进行试验;若吸湿增重5%以下,且其他考察项目符合要求,则不再进行此项试验。
恒湿条件可通过在密闭容器如干燥器下部放置饱和盐溶液实现,根据不同相对湿度的要求,选择NaCl饱和溶液(15.5~60℃,相对湿度75%±1%)或KNO3饱和溶液(25℃,相对湿92.5%)。
3.低温试验***药物置适宜的密封洁净容器中,0℃温度下放置10天,于第5天和第10天取样,按***药物成品质量标准进行全检。
稳定性试验规定,稳定性指导原则,稳定性试验方法
FDA药物稳定性试验指导原那么药品稳定性试验规定每年底制定下年度原料和制剂成品稳定性试验书面方案,内容包括:规格标准、检验方法、检验周期、每批数量、考察工程、考察频次、时间等。
经批准后执行,新开发的制剂品种在开发阶段应制定稳定性方案。
3公司药品生产用原料稳定性试验可采用影响因素试验法:3.1将一批供试品除去包装以后,平放在平皿中,在以下条件下按规定贮存,检测重点考察工程各项质量指标的变化情况。
重点考察工程包括:性状、熔点、含量、有关物质、吸湿性及根据药品性质选定的考察工程。
影响因素试验条件:3.1.1暴露在常温空气中;3.1.2高温试验,温度分别为60℃、40℃两个温度水平;3.1.3高湿试验,湿度分别为90%±5%、75%±5%两个湿度水平;3.1.4强光照射试验,照度为4500LX±500LX4制剂稳定性试验:4.1加速试验:取供试品三批,按市售包装,在温度40℃±2℃,相对温度75%±5%的条件下放置6 个月,在第一个月、第二个月、第三个月、第六个月末取样检测各剂型规定的重点考察工程的质量指标变化情况。
片剂的重点考察工程为:性状、含量、有关物质、崩解时限或溶出度。
硬胶囊剂的重点考察工程为:外观、内容物色泽、含量、有关物质、崩解时限或溶出度、水份。
液体制剂的重点考察工程为:性状、相对密度、含量、pH值、微生物限度检查。
3个月后测试符合要求,有效期暂定为2年,6个月后测试符合要求有效期暂定为3年。
4.2长期试验:取供试品三批,按市售包装,在规定保存条件下贮存,每年检测一次,重点考察工程的质量指标变化情况,观察3年的检验结果,以确定产品的贮存期或有效期。
5严格按照批准的书面稳定性方案,做好试验记录,如发现异常情况,采取措施及时调整。
6试验完毕后,对试验结果进展数理统计后处理,评定并作出结论。
写出稳定性试验报告,所有资料归档保存。
留样观察管理制度留样的环境及要求根据本公司生产的品种的贮存需用,设专用的留样观察室,分为常温区、阴凉区,留样室要求避光、枯燥、通风、防虫鼠。
产品稳定性试验报告
产品稳定性试验报告一、试验概述本次试验旨在评估产品的稳定性,并检测其在不同环境条件下的表现。
试验包括以下内容:1. 温度试验:将产品暴露在不同温度条件下,观察其性能和功能是否受影响。
2. 湿度试验:将产品暴露在不同湿度条件下,评估其防潮性能和耐湿度能力。
3. 震动试验:通过模拟运输过程中的震动情况,检测产品的抗震能力和结构稳定性。
4. 可靠性试验:通过长时间连续运行产品,评估其稳定性和寿命表现。
二、试验结果1. 温度试验在不同的温度条件下,产品的性能和功能经过测试都保持正常。
无论是低温还是高温环境,产品仍然能够正常工作,没有出现异常情况。
2. 湿度试验在不同湿度条件下,产品经过测试显示出较强的防潮性能和耐湿度能力。
产品的外观和功能没有受到湿度的影响,继续保持正常工作。
3. 震动试验经过模拟的运输过程中的震动试验,产品表现出出色的抗震能力和结构稳定性。
无论在强烈震动的情况下,产品仍然能够保持正常工作,没有出现损坏或功能失效的情况。
4. 可靠性试验长时间连续运行试验结果显示,产品具有良好的稳定性和长寿命表现。
在持续运行的过程中,产品没有出现异常情况,并且保持了正常的功能和性能。
三、结论根据以上试验结果,我们可以得出以下结论:1. 产品具有良好的稳定性,能够在不同的环境条件下正常工作。
2. 产品具有较强的防潮性能和耐湿度能力,适用于湿度较高的环境。
3. 产品具有出色的抗震能力和结构稳定性,适用于运输过程中的震动环境。
4. 产品具有良好的可靠性和长寿命表现,能够持续运行而不受损坏或性能下降。
综上所述,产品稳定性试验结果显示,产品已经通过了各项试验,并且符合相关的稳定性要求。
产品具备良好的性能和可靠性,可以放心使用。
以上报告仅基于试验结果,不涉及实际产品使用情况。
如有其他问题或需要进一步了解,请及时联系我们。
稳定性试验方案
稳定性试验⽅案Stability Study Protocol for Exhibit Batch of Chloroquine Phosphate Tablets USP, 250mg 规格为250 mg的USP磷酸氯喹⽚长期、中期及加速稳定性研究⽅案Prepared By: Date:起草者:⽇期:Reviewed By QA: Date:审核者:⽇期:Approved By: Date:批准者:⽇期:Starting Date: Completed Date:开始⽇期:结束⽇期:Contents⽬录1. Purpose⽬的 (1)2. Scope范围 (1)3. References参考资料 (1)4. General Information基本信息 (1)4.1 Stability Samples稳定性研究样品 (1)4.2 Product Outline样品概述 (2)4.3 Formulation处⽅ (2)4.4 Container-Closure Systems包装 (3)4.5 Labeling标签 (3)4.6 Samples and Package样品与包装 (4)5. Stability Testing稳定性测试 (4)5.1 Sample Receipt and Storage样品接收与储存 (4)5.2 Storage Conditions and Testing Time Points储存条件与检测时间点 (4)5.3 Sampling取样 (5)5.4 Testing Matrix稳定性测试项⽬表 (6)5.5 Parameters and Acceptance Criteria检测项⽬及质量标准 (6)5.6 Degradation products降解产物 (7)6. Data Presentation数据汇总 (7)7. Reporting报告 (7)7.1 Intermediate Reports中期报告 (7)7.2 Summary Report总结报告 (7)7.3 Stability Documents稳定性⽂件夹 (7)8. Appendix附件 (8)1.Purpose⽬的The purpose of stability testing is to provide evidence of how the Quality, Strength, Degradation Products and Purity of the Chloroquine Phosphate Tablets USP, 250mg will change with time under the influence of environmental room temperature and relative humidity conditions. Data collected from the stability study will enable recommended storage conditions and provide justification for establishing and submitting the data to regulatory authorities for approving the shelf life for marketing purposes.In addition, 3 months of the stability data will be submitted to US FDA as required for submission purposes of the ANDA application.此稳定性研究的⽬的是为了考察磷酸氯喹⽚在环境因素的影响下(例如:温度和湿度)其性质、规格、降解产物和含量等随时间⽽变化的规律,依据稳定性研究的数据确定该产品的储藏条件和有效期。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
产品稳定性试验方案
1、目的
考察样品在不同环境条件下(如温度、相对湿度等)的感官、化学、物理及生物学随时间增加其变化程度和规律,从而判断样品包装、贮存条件和保质期内的稳定性。
2、参考资料
食品药品监管总局办公厅关于印发保健食品稳定性试验指导原则的通知
食品储存期加速测试及其应用蔡燕芬佳百欧洲食品(惠州)有限公司516005
3、试验要求
3.1、样品类型粉体类、液体类
3.2 、试验条件
并以3M胶封口包装。
3.4、试验仪器恒温恒湿装置
公司的产品种类较多,试验时只能分批次来进行。
(后附恒温恒湿箱的各种型号及市场价格)
2、仪器工作室的选择,由于试验有加速试验和长期试验,试验的时间较长,建议选购3个工作室的仪器较适合。
5、考察指标与检测方法应按照产品质量标准规定的方法,对样品的卫生学及其与产品质量有关的指标在保质期内的变化情况进行的检测。
6、数据记录、汇总
如有侵权请联系告知删除,感谢你们的配合!
如有侵权请联系告知删除,感谢你们的配合!。