MASCOT介绍与蛋白质鉴定

講員: 蔡沛倫
鎂陞科技股份有限公司Mass Solutions Technology
時間: 2010年7月22日
地點: 台灣大學
MASCOT 介紹與蛋白質鑑定的實際應用
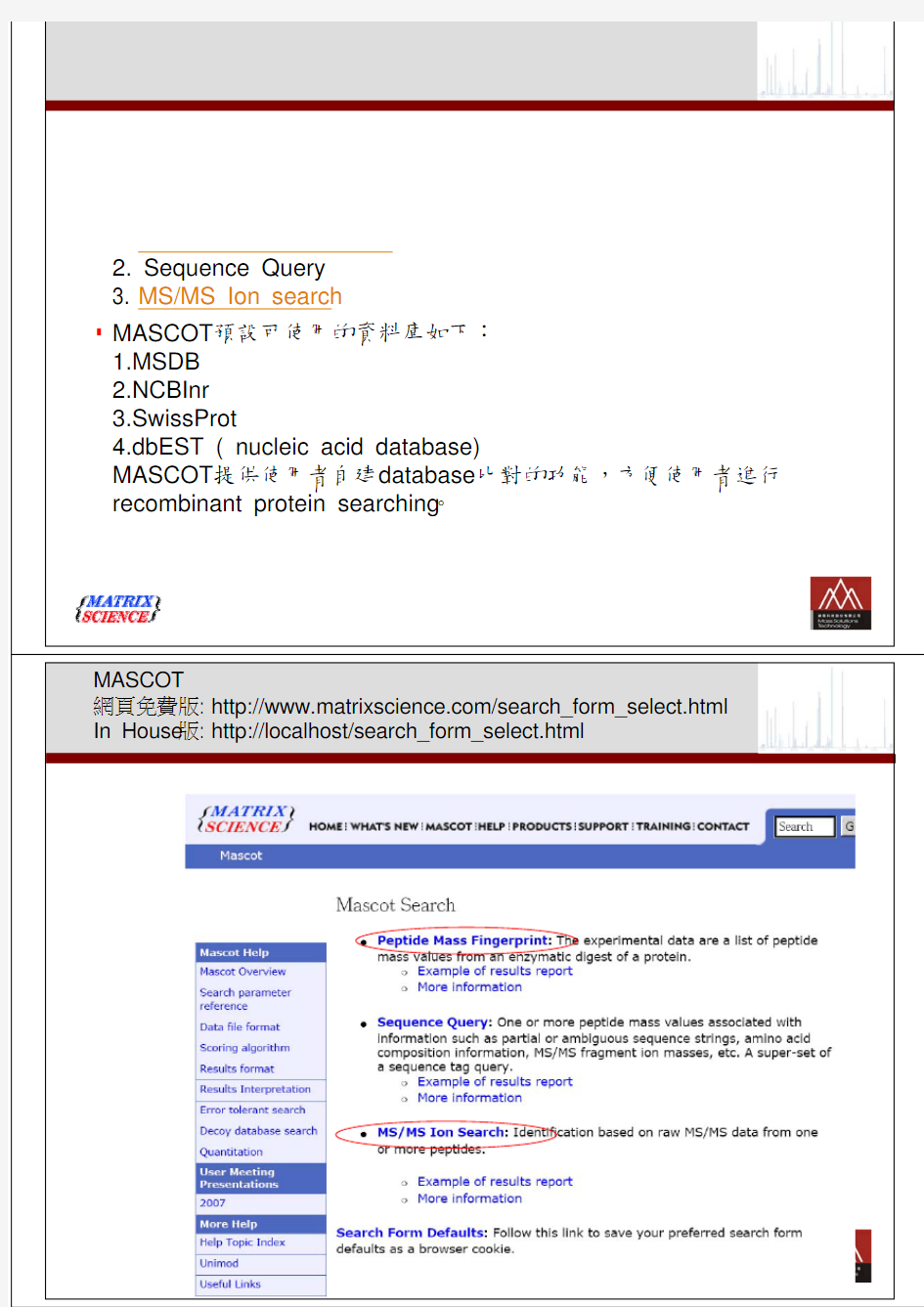
MASCOT
網頁免費版: https://www.360docs.net/doc/4f15418319.html,/search_form_select.html In House版: http://localhost/search_form_select.html
Protein separation Enzymatic digestion Peptide separation
Mass spectrometry Database search
Current Opinion in Chemical Biology2000
1108.53 Peak List
1202.66
1429.59
1731.90
1732.78
1918.13
1919.05
1920.13
2041.99
2185.16
2186.15
2206.10
2207.09
2208.09
2209.24
2210.24
2223.16
2476.24
2499.41
2500.29
2501.34
….
….
….
Database Example
>gi|386828|gb|AAA59172.1| insulin [Homo sapiens]
MALWMRLLPLLALLALWGPDPAAAFVNQHLCGSHLVEALYLVCGERGFFYTPK TRREAEDLQVGQVELGGGPGAGSLQPLALEGSLQKRGIVEQCCTSICSLYQLE NYCN
MS/MS data processing and
database searching
MASCOT results Waters
charge
intensity
m/z
intensity
Fragment ion m/z
Each MSMS (each query
number) has an
individual corresponding
table
Bold: the first time a particular match appears in the report Red: the first ranking peptide match appears
Export search result
For the publication guideline
PTM: p
Error tolerant search
點選後可以看decoy
database比對的結果
以這張結果為例,利用decoy database進
行比對分數達homology的false-postive
rate為4.05%,比對分數達identity的false-
postive rate為0%
... and lots more
蛋白质组学技术在微生物鉴定中的应用-cxr
基于质谱的蛋白质组学技术在微生物鉴定中的应用 Application of Proteomics based on Mass Spectrum in the microorganism identification / classification 微生物这类非常微小而又种类繁多的生物与我们的生活息息相关,近20年来,新的传染病不断出现,如传染性非典型肺炎(SARS)、艾滋病(HIV)、军团菌病、莱姆病(Lyme)、埃博拉出血热(Ebola)、拉沙热(Lassa)、O139型霍乱、致病性大肠杆菌O157:H7引起的出血性肠炎、肠弯曲菌肠炎、汉坦病毒、B组轮状病毒腹泻、疯牛病(克-雅氏病)、禽流感等等,这些新传染病的出现严重威胁人类的身体健康,给人类社会带来了难以估量的后果。同时,随着经济贸易的全球化,国际旅游业的飞速发展也加速了一些传染病的全球化进程,加快了新发传染病的传播速度,也使一些过去得到控制的传染病如结核、多抗药性的链球菌属感染等重新蔓延。当然,除过病原菌以外,腐败菌、有益菌及环境微生物等与我们的健康和生活亦密切相关,这种微生物的多样性为全球制药产业、环境治理、食品工业以及生物技术的发展提供了丰富的资源储备,同时也对人类的健康构成极大的威胁,所以快速、准确鉴定微生物日益成为临床、环境和工业领域的迫切需要,各个国家从来都是不遗余力的在建立、健全菌种资源保存库的同时,积极研究开发快速、准确鉴定微生物身份的新技术和新方法,致力于建立健全微生物资源保存库和鉴定标准库,在临床上为传染病的快速筛查、检测、分离、鉴定、追踪、预警、治疗和预后具有重要意义。目前,尽管微生物鉴定系统实现了鉴定过程的规范化和程序化,将微生物对底物的生化类型与已建立数据库类型相比较来鉴定微生物,但其反应准确性受接种物浓度、孵育条件和试验解释等的影响。自20世纪80~90年代以来,微生物鉴定系统不断发展,自动化程度不断提高,尤其是基于质谱技术的蛋白质组技术和代谢组技术在微生物研究领域的介入,使得微生物鉴定达到了快速、准确、大规模、高通量的水平。 一、微生物鉴定系统方法的发展 (一)微生物鉴定的传统方法 微生物传统的鉴定方法是建立在微生物的形态学、生态学、细胞生理和生化以及基因的基础上的,主要包括以下几类: 1.生化方法 该法检测微生物实际上是测定微生物特异性酶。由于各种微生物所具有的酶系统不完全相同,对许多物质的分解能力亦不一致。因此可利用不同底物产生的不同代谢产物来间接检测该微生物内酶的有无,从而达到检测特定微生物的目的。 2.免疫学技术 免疫学技术是利用特异性抗原抗体反应,观察和研究组织细胞、特定抗原(抗体)的定性
蛋白质结构解析的方法对比综述 (1)
蛋白质结构解析的方法对比综述 工程硕士李瑾 摘要:到目前为止,蛋白质结构解析的方法主要是两种,x射线衍射法和NMR法,这两种方法各有优点和不足。 关键词:x射线衍射法 NMR法 到目前为止,蛋白质结构解析的方法主要是两种,x射线衍射法和NMR法。其中X射线的方法产生的更早,也更加的成熟,解析的数量也更多,第一个解析的蛋白的结构,就是用x晶体衍射的方法解析的。而NMR方法则是在90年代才成熟并发展起来的。这两种方法各有优点和不足[1]。 首先是X射线晶体衍射法。该方法的前提是要得到蛋白质的晶体。通常是将表达目的蛋白的基因经PCR扩增后克隆到一种表达载体中,然后转入大肠杆菌中诱导表达,目的蛋白提纯之后摸索结晶条件,等拿到晶体之后,将晶体进行x射线衍射,收集衍射图谱,通过一系列的计算,得到蛋白质的原子结构[2]。 x射线晶体衍射法的优点是:速度快,通常只要拿到晶体,最快当天就能得出结构,另外不受肽链大小限制,无论是多大分子量的蛋白质或者RNA、DNA,甚至是结合多种小分子的复合体,只要能够结晶就能够得到其原子结构。所以x射线方法解析蛋白的关键是摸索蛋白结晶的条件。该方法得到的是蛋白质分子在晶体状态下的空间结构,这种结构与蛋白质分子在生物细胞内的本来结构有较大的差别。晶体中的蛋白质分子相互间是有规律地、紧密地排列在一起的,运动性较差;而自然界的生物细胞中的蛋白质分子则是处于一种溶液状态,周围是水分子和其他的生物分子,具有很好的运动性。而且,有些蛋白质只能稳定地存在于溶液状态,无法结晶[2]。 核磁共振NMR(nuclear magnetic resonance)现象很早就被科研人员观察到了,但将这种方法用来解析蛋白质结构,却是近一二十年的事情。NMR法具体原理是对水溶液中的蛋白质样品测定一系列不同的二维核磁共振图谱,然后根据已确定的蛋白质分子的一级结构,通过对各种二维核磁共振图谱的比较和解析,在图谱上找到各个序列号氨基酸上的各种氢原子所对应的峰。有了这些被指认的峰,就可以根据这些峰在核磁共振谱图上所呈现的相互之间的关系得到它们所对应的氢原子之间的距离。[3]可以想象,正是因为蛋白质分子具有空间结构,在序列上相差甚远的两个氨基酸有可能在空间距离上是很近的,它们所含的氢原子所对应的NMR峰之间就会有相关信号出现[4] 。通常,如果两个氢原子之间距离小于0.5纳米的话,它们之间就会有相关信号出现。一个由几十个氨基酸残基组成的蛋白质分子可以得到几百个甚至几千个这样与距离有关的信号,按照信号的强弱把它们转换成对应的氢原子之间的距离,然后运用计算机程序根据所得到的距离条件模拟出该蛋白质分子的空间结构。该结构既要满足从核磁共振图谱上得到的所有距离条件,还要满足化学上有关原子与原子结合的一些基本限制条件,如原子间的化学键长、键角和原子半径等[4]。 NMR解析蛋白结构常规步骤如下:首先通过基因工程的方法,得到提纯的目的蛋白,在蛋白质稳定的条件下,将未聚合,而且折叠良好的蛋白样品(通常是1mM-3mM,500ul,PH6-7的PBS)装入核磁管中,放入核磁谱仪中,然后由写好的程序控制谱仪,发出一系列的电磁波,激发蛋白中的H、13N、13C原子,等电磁波发射完毕,再收集受激发的原子所放出的“能量”,通过收集数据、谱图处理、电脑计算从而得到蛋白的原子结构[5] [6]。 用NMR研究蛋白质结构的方法,可以在溶液状态进行研究,得到的是蛋白质分子在溶液中的结构,这更接近于蛋白质在生物细胞中的自然状态[7]。此外,通过改变溶液的性质,还可以模拟出生物细胞内的各种生理条件,即蛋白质分子所处的各种环境,以观察这些周围环境的变化对蛋白质分子空间结构的影响。在溶液环境中,蛋白质分子具有与自然环境中类
蛋白质分离纯化的步骤
蛋白质分离纯化的一般程序可分为以下几个步骤: (一)材料的预处理及细胞破碎 分离提纯某一种蛋白质时,首先要把蛋白质从组织或细胞中释放出来并保持原来的天然状态,不丧失活性。所以要采用适当的方法将组织和细胞破碎。常用的破碎组织细胞的方法有: 1. 机械破碎法 这种方法是利用机械力的剪切作用,使细胞破碎。常用设备有,高速组织捣碎机、匀浆器、研钵等。 2. 渗透破碎法 这种方法是在低渗条件使细胞溶胀而破碎。 3. 反复冻融法 生物组织经冻结后,细胞内液结冰膨胀而使细胞胀破。这种方法简单方便,但要注意那些对温度变化敏感的蛋白质不宜采用此法。 4. 超声波法 使用超声波震荡器使细胞膜上所受张力不均而使细胞破碎。 5. 酶法 如用溶菌酶破坏微生物细胞等。 (二)蛋白质的抽提 通常选择适当的缓冲液溶剂把蛋白质提取出来。抽提所用缓冲液的pH、离子强度、组成成分等条件的选择应根据欲制备的蛋白质的性质而定。如膜蛋白的抽提,抽提缓冲液中一般要加入表面活性剂(十二烷基磺酸钠、tritonX-100 等),使膜结构破坏,利于蛋白质与膜分离。在抽提过程中,应注意温度,避免剧烈搅拌等,以防止蛋白质的变性。(三)蛋白质粗制品的获得选用适当的方法将所要的蛋白质与其它杂蛋白分离开来。比较方便的有效方法是根据蛋白质溶解度的差异进行的分离。常用的有下列几种方法: 1.等电点沉淀法不同蛋白质的等电点不同,可用等电点沉淀法使它们相互分离。 2.盐析法 不同蛋白质盐析所需要的盐饱和度不同,所以可通过调节盐浓度将目的蛋白沉淀析出。被盐析沉淀下来的蛋白质仍保持其天然性质,并能再度溶解而不变性。 3.有机溶剂沉淀法 中性有机溶剂如乙醇、丙酮,它们的介电常数比水低。能使大多数球状蛋白质在水溶液中的溶解度降低,进而从溶液中沉淀出来,因此可用来沉淀蛋白质。此外,有机溶剂会破坏蛋白质表面的水化层,促使蛋白质分子变得不稳定而析出。由于有机溶剂会使蛋白质变性,使用该法时,要注意在低温下操作,选择合适的有机溶剂浓度。 (四)样品的进一步分离纯化
蛋白质质谱鉴定
广州辉骏生物科技有限公司 蛋白质质谱鉴定 一、技术概述 质谱是将待测物质变为气态离子并将离子按质荷比(m/z)进行分离,检测各种离子谱峰的强度而实现分析的一种方法。 蛋白质定性通常采用质谱分析结合数据库检索的方法,所分析的样本可以是蛋白质溶液、蛋白质胶条或胶点。 简单蛋白样本,例如双向电泳斑点或纯化蛋白,通常采用MALDI-TOF/TOF质谱(MS/MS)进行分析。 混合蛋白样本,例如蛋白溶液,或SDS-PAGE条带,通常采用液相色谱-质谱联用(LC-MS/MS)技术进行分析。应用领域有:亚细胞组分的全谱分析,IP、co-IP、Pull-down后的互作蛋白鉴定,或其他中等复杂蛋白样本的鉴定。 二、技术原理 串联质谱(MS/MS)检测蛋白的原理是:蛋白先经胰酶消化成肽段,肽段在质谱仪中离子化后,会带上一定量的电荷,通过检测器分析,可得到各肽段的质荷比(m/z),从而得知各肽段的相对分子质量。为获得肽段的序列信息,质谱仪会选取某些肽段进行破碎,再次分析,获得二级质谱。用检索软件选择相应的数据库对质谱数据进行分析,同时以打分的形式评判鉴定结果,当打分大于某个阈值时,即判定质谱鉴定成功,反之则鉴定失败。 LC-MS/MS方法是将蛋白酶切消化为肽段混合物,之后这些肽段先经高效液相色谱分离形成简单的组分,再进行串联质谱(MS/MS)分析;因此适合于混合蛋白样本的鉴定。 三、技术优势 1. 采用高效液相色谱和质谱联用的分析方法,可以一次性鉴定成百上千种蛋白质。 2. 鉴定准确性和灵敏度高。 四、技术流程 蛋白样本制备——蛋白酶解——串联质谱分析(或LC-MS/MS分析)——数据库检索——蛋白质鉴定结果
蛋白质结构分析原理及工具-文献综述
蛋白质结构分析原理及工具 (南京农业大学生命科学学院生命基地111班) 摘要:本文主要从相似性检测、一级结构、二级结构、三维结构、跨膜域等方面从原理到方法再到工具,系统地介绍了蛋白质结构分析的常用方法。文章侧重于工具的列举,并没有对原理和方法做详细的介绍。文章还列举了蛋白质分析中常用的数据库。 关键词:蛋白质;结构预测;跨膜域;保守结构域 1 蛋白质相似性检测 蛋白质数据库。由一个物种分化而来的不同序列倾向于有相似的结构和功能。物种分化后形成的同源序列称直系同源,它们通常具有相似的功能;由基因复制而来的序列称为旁系同源,它们通常有不同的功能[1]。因此,推测全新蛋白质功能的第一步是将它的序列与进化上相关的已知结构和功能的蛋白质序列比较。表一列出了常用的蛋白质序列数据库和它们的特点。 表一常用蛋白质数据库 网址可能有更新 氨基酸替代模型。进化过程中,一种氨基酸残基会有向另一种氨基酸残基变化的倾向。氨基酸替代模型可用来估计氨基酸替换的速率。目前常用的替代模型有Point Accepted Mutation (PAM)矩阵、BLOck SUbstitution Matrix (BLOSUM)矩阵[2]、JTT模型[3]。 序列相似性搜索工具。序列相似性搜索又分为成对序列相似性搜索和多序列相似性搜索。成对序列相似性搜索通过搜索序列数据库从而找到与查询序列相似的序列。分为局部联配和全局联配。常用的局部联配工具有BLAST和SSEARCH,它们使用了Smith-Waterman 算法。全局联配工具有FASTA和GGSEARCH,基于Needleman-Wunsch算法。多序列相似性搜索常用于构建系统发育树,这里不阐述。表二列举了常用的成对序列相似性比对搜索工具
SDS-PAGE测定蛋白质分子量及蛋白质的纯度鉴定
SDS-PAGE测定蛋白质分子量及蛋白质的纯度鉴定一、实验目的与原理 蛋白质在聚丙烯酰胺凝胶中电泳时,它的迁移取决于它所带电荷以及分子大小和形状等因素。1967年Shapiro等人发现,如果在聚丙烯酰胺系统中加入阴离子去污剂十二烷基磺酸钠(SDS),大多数蛋白质能与SDS按一定比例结合,即每克蛋白质结合1.4g的SDS-复合物都带上相同密度的负电荷,它的量大大超过了蛋白质分子原有的电荷量,因而消除了蛋白质原有的电荷差别,使蛋白质分子电泳的迁移率主要取决于本身的分子量,而与蛋白质所带的电荷无关,在一定条件下,蛋白质的分子量的对数与电泳迁移率间呈负相关。 本实验的目的是对多酚氧化酶的纯化度鉴定及分子量的测定,通过实验,学习和掌握SDS聚丙烯酰胺凝胶电泳法蛋白质纯度和分子量的鉴定。 二、仪器与试剂 1、材料: 硫酸铵盐析沉淀的多酚氧化酶粗酶样品、DEAE-纤维素DE52柱层析的样品,Sephadex G-100柱层析的样品。 2、试剂: (1)丙稀酰胺(Acr母液):30%Acr (Acr/Bis) (2)10%的SDS溶液 (3)10%的过硫酸铵溶液 (4)四甲基乙二胺(TEMED) (5)分离胶缓冲液:1.5M Tris,PH8.8 (6)浓缩胶缓冲液:1.0M Tris,PH6.8 (7)电极缓冲液:10×30g Tris,125g 甘氨酸和5g SDS,加水溶解定容至1000ml,pH8.3 (8)样品缓冲液:0.2M Tris,PH6.8,1%SDS,30%甘油,巯基乙醇及溴酚兰 (9)染色液:0.15%考马斯亮蓝R250,溶于脱色液
(10)脱色液:50%的甲醇,7%的冰醋酸的水溶液 (11)标准分子量蛋白。 3、仪器设备: 电泳仪、垂直电泳槽等。 三、操作步骤 1、凝胶制备: 用两块电泳玻璃板制成垂直板槽(不能漏胶),垂直放置。将配制好的分离胶溶液倒入,滴加入无离子水,待凝胶聚集后,倒出无离子水,用吸水纸吸干,倒入浓缩胶,再插入梳子。 2、上样: 分别取样品若干ml于离心管中,按1/1~1/5比例加入5×样品缓冲液,再沸水浴中加热3~5min,取出待用。用微量注射器分别吸取不超过30μl不同浓度的标准蛋白样品和试验样品注入样品槽。点样结束后,调节电泳仪电流到10mA(2~3mA/em),保持电流稳定不变,当溴酚蓝迁移到离分离胶底1~2cm时,即可停止电泳。 3、染色: 电泳完毕后,取出凝胶板,浸入染色液中,在37℃温箱中保温过夜。倒掉染色液,24h后,即可看到清晰的蛋白质条带。 四、结果(略) 五、注意事项 1、SDS与蛋白质的结合按质量成比例(即:1.4gSDS/g蛋白质),如果比例不当,就不能得到准确的数据。 2、用SDS-聚丙烯酰胺凝胶电泳法测定蛋白质相对分子量时,必须同时作标准曲线。不能利用这次的标准曲线作为下次用。 3、有些蛋白质由亚基(如血红蛋白)或两条以上肽链(α-胰凝乳蛋白酶)组成的,它们在巯基乙醇和SDS的作用下解离成亚基或多条单肽链。因此,对于这一类蛋白质,SDS-聚丙烯酰胺凝胶电泳法测定的只是它们的亚基或是单条肽链的相对分子量。 4、有的蛋白质(如:电荷异常或结构异常的蛋白质;带有较大辅基的蛋白质)不能采用该法测相对分子量。 5、如果该电泳中出现拖尾、染色带的背景不清晰等现象,可能是SDS不纯引起。
蛋白质组学复习题
蛋白质组:一种细胞、组织或完整的生物体所拥有的全套蛋白质 蛋白质组学:应用各种技术手段来研究蛋白质组的一门新兴学科,即研究细胞在不同生理或病理条件下蛋白质表达的异同,对相关蛋白质进行分类和鉴定。更重要的是蛋白质组学的研究要分析蛋白质间相互作用和蛋白质的功能 F-2D-DIGE荧光差异显示双向凝胶电泳 在传统双向凝胶电泳基础上发展起来的定量分析凝胶上蛋白质点的新方法 具体操作步骤: 首先将待比较的两种样品提取的总蛋白分别与两种不同的荧光标记试剂(Cy2, C y3或Cy5)进行共价标记,然后将不同荧光染料标记的两种待比较的蛋白质样品等量混合,上样进行双向电泳,2D凝胶在成像仪上用两种不同波长激发,并将两种样品的荧光图谱显示成像,用2D分析软件进行定量分析。将显示有明显差异的蛋白点切下后进行鉴定分析,从而确定差异表达的蛋白质 PF-2D:以蛋白质组或复杂的蛋白质混合物为分离对象,通过一维色谱聚焦(即将复杂蛋白质混合物按蛋白质的等电点差异进行分离)和二维无孔硅胶反相HPLC分离色谱(即按蛋白质的疏水性差异进行分离)模式进行分级分离,软件将液相色谱的分离图转换为双向电泳形式,一维为等电聚焦结果,二维为分子量分离结果,为蛋白质组学研究提供了一个创新的技术平台。 从头测序(Do novo sequencing):不使用来自蛋白质数据库的信息,直接解释串联质谱数据的方法和技术 通过20种氨基酸残基的质量等信息以及主要类型离子谱峰间的质荷比(M/Z)差值来构造序列。这类蛋白质鉴定方法就称为蛋白质的从头测序。 Tandem mass spectrometry串联质谱:由2台质谱仪经1个碰撞室串联而成。首先利用第一台质谱仪选择特殊离子,在进入第二台质谱仪之前,先进入碰撞室与加入其中的气体(氮气或氦气)产生碰撞形成各种离子,再利用第二台质谱仪依各离子的质核比不同而分离,形成第二次质谱,可作为离子结构判断的依据 肽质量指纹图谱(peptide mass fingerprint)PMF:指特定的蛋白酶对蛋白质进行酶解后的质谱分析得到一套多肽质量图谱,这种特性就象指纹一样,每种蛋白都具有特定的质量肽谱。碰撞诱导解离(CID):在三级四级杆质谱中,第一级Q1和第三级Q3四级杆是质量过滤器,第二级四级杆Q2仅施加射频电压,充当产生碎片离子的碰撞室,从Q1传送来的肽离子在碰撞室内经惰性气体如Ar和N2的碰撞诱导产生正离子,这一过程称为碰撞诱导解离 肽序列标签(Peptide sequence tag, PST)技术 蛋白质由20种氨基酸组成,5~6个氨基酸残基的序列片段在一个蛋白质组成中具有很高的特异性,这个片段称为PST,可用于蛋白质鉴定。源后衰变(PSD):母离子在源内离子化后,在飞行过程中会产生一系列的亚稳离子,恰这些离子主要是沿多肽骨架断裂形成,产生的这些碎片离子在理论上包含着这一肽段的氨基酸序列信息。由于这一过程发生在源内离子化之后,所以称为源后衰变。 蛋白质芯片:通过微加工和微电子技术对固相载体进行特殊的化学处理,将大量已知的蛋白质如抗原、抗体、受体、配体等有序的固定在载体上,使其与待检蛋白质分子进行特异性结合或反应,经洗涤和检测信号的强弱判断样品中的靶分子数量和特征,从而实现对蛋白质组分的准确、快速和大信息量的筛检。 蛋白质组样品的全息制备: 就是要求样品制备尽可能获得所有的蛋白质,但由于蛋白质种类多、丰度不一和物理化学特性不同等,要达到真正的全息制备是有难度的. 蛋白质组学的主要技术,简述它们的基本原理 双向电泳质谱酵母双杂交噬菌体显示技术蛋白质芯片技术荧光差异显示双向电泳同位素亲和标签生物信息学色谱-质谱联用Edman 降解法
蛋白质提取、纯化、鉴定的方法(二)
蛋白质提取、纯化、鉴定的方法(二) 一、层析技术 1.离子交换层析的亲和洗脱这种技术结合了离子交换与亲和层析。如在某一pH时,目的蛋白质带正(负)电荷,用阳(阴)离子交换剂吸附,这一过程去除了很大一部分不吸附的杂蛋自。然后用该目的蛋白质的配体来洗脱,该配体特异性地结合目的蛋白质并使之洗脱,但不洗脱其他吸附的蛋白质,达到纯化的目的。注意,该配体需带有一定量的阴(阳)电荷,有效降低目的蛋白质与阳(阴)离子交换剂之间的电荷相互作用。 2.固相金属亲和层析重组蛋白质可在C-或N-端引入组氨酸标签,一般为6个组氨酸残基(His-tag)。这些组氨酸残基与过渡金属(transitionalmetals)Ni2+或Co2+形成配位键。用固相化的Ni2+或Co2+(如商品化的树脂,Ni-NTA)可吸附带有His-tag的重组蛋白质,用含有咪唑(imidazole)的缓冲液可洗脱重组蛋白质。注意,有些含有较多组氨酸的蛋白质也可与吸附剂结台,但较弱,因此可用低浓度的咪唑洗脱;在层析过程中不能引入金属螯合剂如EDTA;避免使用还原剂如DTT或DTE,但可用低浓度的巯基乙醇。 该技术也用于提取磷酸化的蛋白质。将螫合剂交联到树脂,螯合三价铁或三价镓,该亲和吸附剂可吸附混合物中的磷酸化的蛋白质。洗去不吸附的非磷酸化蛋白质后,用磷酸缓冲液即可将磷酸化蛋白质从该亲和吸附剂上洗脱。要注意的是酸性蛋白质也可被不同程度地吸附。 3.凝胶过滤该技术过去也被称为分子筛。构成凝胶的小珠(bead)中有大小不一的孔,分子量大的分子能进入较大的孔而不能进入小的孔,分子量小的则不仅能进入较大的孔也能进入小的孔,因此在层析过程中,小分子经过的路程较长而大分子经过的路程较短,如此就可分离分子量不同的蛋白质。然而,分子量相近的蛋白质非常多,因此,用这种技术得到的蛋白质是分子量相近的混合蛋白质。然而这种技术在某些研究中很有用,如丙酮酸激酶M2(PKM2)由四个相同的亚基组成,PKM2在细胞中以三种形式存在——单体、二聚体、四聚体,这三种形式的功能不同,若要鉴定细胞中PKM2的各种形式的量,先用凝胶过滤技术分离细胞裂解液中的PKM2的三种形式,之后用Western blot对每一种形式的PKM2做相对定量。 4.反相层析该技术是指用疏水固相的一种层析技术。“反相”是相对“正相”而言,正相是指亲水的固相如硅胶表面带有硅羟基(silanol group),硅羟基可与被分离的化台物相互作用,被分离的化合物的亲水性越强,则滞留在正相
蛋白质的鉴定
蛋白质的鉴定 【实验一】生物组织中还原糖、脂肪、蛋白质的鉴定 一、教学目的 初步掌握鉴定生物组织中还原糖、脂肪、蛋白质的基本方法。 二、教学建议 教材中本实验安排为验证性实验,有条件的学校可以改为探索性实验,安排在讲课之前,或与讲课同步进行。 本实验难度并不大,但内容较多,实验时间较长,因此,必须作周密安排,才能按时完成。实验中应注意以下几点。 1.增设教师演示实验。上课之前,教师应该准备好做演示实验所需的实验材料、用具、仪器和试剂等。同时,逐项完成还原糖、脂肪、蛋白质3类有机物的鉴定实验。在实验课上,将3个实验的正确结果分别展示在讲台上,并作扼要的介绍,以便使学生将自己的实验结果与教师的演示实验作比较。 2.实验中学生应分工合作。在“还原糖的鉴定”实验中,当每组两个学生中的一个制备生物组织样液时,另一个学生可以用酒精灯将水煮开,以便缩短实验的等待时间。在“脂肪的鉴定”实验中,一个学生制作临时装片时,另一个学生则可以调试显微镜。另外,在完成前两个实验时,一个学生
洗刷试管、清洗玻片和整理显微镜,另一个学生则可以进行后一个实验的操作。 3.关于鉴定还原糖的实验,在加热试管中的溶液时,应该用试管夹夹住试管上部,并放入盛开水的大烧杯中加热。注意试管底部不要接触烧杯底部,同时试管口不要朝向实验者,以免试管内溶液沸腾时冲出试管,造成烫伤。如果试管内溶液过于沸腾,可以上提试管夹,使试管底部离开大烧杯中的开水。 4.做鉴定还原糖和蛋白质的实验时,在鉴定之前,可以留出一部分样液,以便与鉴定后的样液的颜色变化作对比,这样可以增强说服力。 5.斐林试剂的甲液和乙液混合均匀后方可使用,切勿将甲液和乙液分别加入组织样液中。 三、参考资料 还原糖的鉴定原理生物组织中普遍存在的还原糖种类较多,常见的有葡萄糖、果糖、麦芽糖。它们的分子内都含有还原性基团(游离醛基或游离酮基),因此叫做还原糖。蔗糖的分子内没有游离的半缩醛羟基,因此叫做非还原性糖,不具有还原性。本实验中,用斐林试剂只能检验生物组织中还原糖存在与否,而不能鉴定非还原性糖。 斐林试剂由质量浓度为0.1 g/L的氢氧化钠溶液和质量浓度为0.05 g/L的硫酸铜溶液配制而成,二者混合后,
MASCOT介绍与蛋白质鉴定
講員: 蔡沛倫 鎂陞科技股份有限公司Mass Solutions Technology 時間: 2010年7月22日 地點: 台灣大學 MASCOT 介紹與蛋白質鑑定的實際應用
MASCOT 網頁免費版: https://www.360docs.net/doc/4f15418319.html,/search_form_select.html In House版: http://localhost/search_form_select.html
Protein separation Enzymatic digestion Peptide separation Mass spectrometry Database search Current Opinion in Chemical Biology2000
1108.53 Peak List 1202.66 1429.59 1731.90 1732.78 1918.13 1919.05 1920.13 2041.99 2185.16 2186.15 2206.10 2207.09 2208.09 2209.24 2210.24 2223.16 2476.24 2499.41 2500.29 2501.34 …. …. ….
Database Example >gi|386828|gb|AAA59172.1| insulin [Homo sapiens] MALWMRLLPLLALLALWGPDPAAAFVNQHLCGSHLVEALYLVCGERGFFYTPK TRREAEDLQVGQVELGGGPGAGSLQPLALEGSLQKRGIVEQCCTSICSLYQLE NYCN
蛋白质组学技术服务
2D 电泳及质谱鉴定技术服务 ----生工带您走进蛋白质组学时代 ? 实验原理: 1、 双向凝胶电泳是将不同种类的蛋白质按照等电点和分子量差异进行高分辨率分离的分析方法。成功的二维电泳可以将2000到3000种蛋白质进行分离,是目前唯一的同时能分离成百上千种蛋白质的工具。通过对蛋白质双向电泳的图谱扫描,用相关软件(ImageMaster 等)进行图谱差异分析,找到差别点。然后把差异点切出,进行脱色、酶解。最后样品进入质谱测试,得到质谱原始数据文件。通过搜库可得到差异蛋白质的详细信息。 2、 ABI 4800 串联质谱(MALDI-TOF-TOF-MS )是目前对SDS-PAGE 胶上的蛋白条带和2D 胶上差异点蛋白,切取并经过酶解后进行鉴定最常用的质谱,它在肽指纹图谱(一级质谱)的基础上选择强度最大的10个峰进行进行二级质谱,对肽段碎片的分子量精确测定,再将一级和二级质谱数据整合并使用GPS 3.6(Applied Biosystems )和Mascot2.1(Matrix Science)对质谱数据进行分析和蛋白鉴定。其提供的专一性信息丰富,对于数据库的检索,特别是对数据量丰富、冗余信息多的数据库的检索,其鉴定结果比肽指纹图谱可靠的多,该ABI 4800也可以不经过酶解直接对蛋白进行分子量精确测定 。 ? 主要步骤: 1、 样品制备(蛋白提取):主要包括植物、动物细胞或组织等的破碎、离心以及定量。 2、 第一向IEF 等电聚焦实验。 3、 第二向SDS-PAGE 实验。 4、 硝酸银染色或考马斯亮蓝染色。 5、 图像分析及数据处理。 6、 提供双向电泳的正式实验报告。 7、 选取感兴趣的蛋白点进行酶解。 8、 对酶解后蛋白进行质谱技术分析(Maldi-Tof-Tof/MS )。 9、 提供质谱的正式实验报告。 经典蛋白质组学研究策略
蛋白表达、分离和纯化
蛋白质的表达、分离、纯化和鉴定 来源:易生物实验浏览次数:2704网友评论0 条第一部分蛋白质的表达、分离、纯化克隆基因在细胞中表达对理论研究和实验应用都具有重要的意义。通过表达能探索和研究基因的功能以及基因表达调控的机理,同时克隆基因表达出所编码的蛋白质可供作 结构与功能的研究。 第二部分蛋白质的鉴定电泳可用于分离复杂的蛋白质混合物,研究蛋白质的亚基组成等。在聚丙烯酰胺凝胶电泳中,凝胶的孔径,蛋白质的电荷,大小,性质等因素共同决定了蛋白质的电泳迁移率。 关键词:蛋白质蛋白质表达克隆基因聚丙烯酰胺凝胶电泳氯霉素酰基转移酶十二烷基硫酸钠SDS聚丙烯酰 胺凝胶 第一部分蛋白质的表达、分离、纯化 目的要求 (1)了解克隆基因表达的方法和意义。 (2)了解重组蛋白亲和层析分离纯化的方法。 实验原理 克隆基因在细胞中表达对理论研究和实验应用都具有重要的意义。通过表达能探索和研究基因的功能以及基因表达调控的机理,同时克隆基因表达出所编码的蛋白质可供作结构与功能的研究。大肠杆菌是目前应用最广泛的蛋白质表达系统,其表达外源基因产物的水平远高于其它基因表达系统,表达的目的蛋白量甚至能超过细菌总蛋白量的80%。本实验中,携带有目标蛋白基因的质粒在大肠杆菌BL21中,在37℃,IPTG诱导下,超量表达携带有6个连续组氨酸残基的重组氯霉素酰基转移酶蛋白,该蛋白可用一种通过共价偶连的次氨基三乙酸(NTA)使镍离子(Ni2+)固相化的层析介质加以提纯,实为金属熬合亲和层析(MC AC)。蛋白质的纯化程度可通过聚丙烯酰胺凝胶电泳进行分析。 试剂和器材
一、试剂 [1] LB液体培养基:Trytone 10g, yeast extract 5g, NaCl 10g, 用蒸馏水配至1000mL. [2] 氨苄青霉素:100mg/mL [3] 上样 缓冲液:100 mM NaH2PO4, 10 mM Tris, 8M Urea, 10 mM2-ME, pH8.0 [4] Washing Buffer:100 mM NaH2PO4, 10 mM Tris, 8 M Urea, pH6.3 [5] Elution Buffer:100 mM NaH2PO4, 10 mMTris, 8M Urea, 500 mM Imidazole, pH 8.0 [6] IPTG 易生物仪器库:.ebioe./yp/product-list-42.html 易生物试剂库:.ebioe./yp/product-list-43.html 二、器材 摇床,离心机,层析柱(1′10 cm) 操作方法 一、氯霉素酰基转移酶重组蛋白的诱导 1. 接种含有重组氯霉素酰基转移酶蛋白的大肠杆菌BL21菌株于5mL LB液体培养基中(含100ug/mL 氨苄青霉素),37℃震荡培养过夜。 2. 转接1mL过夜培养物于100mL(含100ug/mL 氨苄青霉素)LB液体培养基中,37℃震荡培养至OD600 = 0.6 - 0.8。取10ul 样品用于SDS-PAGE 分析。 3. 加入IPTG至终浓度0.5 mmol/l, 37℃继续培养1-3h.
蛋白质组学研究的基本步骤
请简述蛋白质组学研究的基本步骤 1.蛋白质样品的制备:蛋白质样品的制备是蛋白质组学研究的首要环节,也是最为重要的部分。蛋白质样品的质量直接影响到科学研究的真实性和可信度。 2.蛋白质的分离:双向凝胶电泳技术是目前最基础和常用的蛋白质分离方法,它能将数千种蛋白质同时分离与展示的分离技术。双向电泳分为等电聚焦电泳和SDS-PAGE两个步骤,即先进行等电聚焦电泳,按照pI的不同将蛋白分离,然后再进行SDS-PAGE按照分子量的大小不同对蛋白进行分离。IPG胶条的应用,大大提高了双向电泳的重复性。 3. 蛋白质双向电泳凝胶的染色。目前双向电泳凝胶的染色的方法有3种,分别为考马斯亮蓝染色法、银染法和荧光染色法。考马斯亮蓝染色法,操作简便,无毒性,染色后的背景及对比度良好,与下游的蛋白质鉴定方法兼容,但灵敏度较低,可以检测到30~100 ng蛋白质。银染法是一种较为流行的染色方法,银染成本较低,灵敏度高,可检测少到2~5ng的蛋白。荧光试剂显色对蛋白质无固定作用,与质谱兼容性好,而其灵敏度与银染相仿,但线性范围要远高于银染,这使二维电泳分离蛋白质的荧光检测受到普遍关注和应用。 4.双向电泳凝胶图像的采集与分析:图像采集系统通过投射扫描根据吸光度的大小获碍蛋白质点的光密度信息。一般来说,该光密度值与蛋白质点的表达丰度成正比,以便于软件分析时的定量比较。完成图像采集后采用ImageMaster等图像分析软件进行分析。分析步骤:蛋白质点检测、背景消减、归一化处理、蛋白质点匹配。 5.蛋白质鉴定:蛋白质鉴定是蛋白质组学研究中的核心内容。目前蛋白质鉴定技术主要有Edman 降解法测序、质谱。质谱是目前最常用的蛋白质鉴定方法。质谱技术的基本原理是带电粒子在磁场或电场中运动的轨迹和速度依粒子的质量与携带电荷之比质荷比( m/z) 的不同而变化,可以据此来判断粒子的质量和特性。质谱完成后利用蛋白质的各种属性参数如相对分子质量、等电点、序列、氨基酸组成、肽质量指纹谱等在蛋白质数据库中检索,寻找与这些参数相符的蛋白质。
实验十蛋白质的表达、分离纯化和鉴定
实验十蛋白质的表达、分离纯化和鉴定 第一部分蛋白质的表达、分离纯化 目的要求 (1)了解重组蛋白表达的方法和意义。 (2)了解重组蛋白亲和层析分离纯化的方法。 实验原理 目的基因在宿主细胞中的高效表达及表达的重组蛋白的分离纯化对理论研究和实验应用都具有重要的意义。通过表达能探索和研究基因的功能以及基因表达调控的机理,同时目的基因表达出所编码的蛋白质可供作结构与功能的研究。大肠杆菌是目前应用最广泛的蛋白质表达系统,其表达外源基因产物的水平远高于其它表达系统,表达的目的蛋白量甚至能超过细菌总蛋白量的80%。本实验中,携带有目标蛋白基因的质粒在大肠杆菌BL21(DE3)中,在37℃,IPTG诱导下,超量表达携带有6个连续组氨酸残基的重组氯霉素酰基转移酶蛋白,该蛋白N端带有6个连续的组氨酸残基,可通过固相化的镍离子(Ni2+)亲和层析介质加以分离纯化,称为金属熬合亲和层析(MCAC)。蛋白质的纯化程度可通过聚丙烯酰胺凝胶电泳进行分析。 试剂和器材 一、试剂 [1] LB液体培养基:Trytone 10g, yeast extract 5g, NaCl 10g, 用蒸馏水配至1000mL。 [2] 氨苄青霉素:100mg/mL。 [3] 上样缓冲液(GLB):100 mM NaH2PO4, 10 mM Tris, 8M Urea, 1 mM β-巯基乙醇, pH8.0。 [4] 清洗缓冲液(UWB):100 mM NaH2PO4, 10 mM Tris, 8 M Urea, pH6.3。 [5] 洗脱液缓冲液:100 mM NaH2PO4, 10 mM Tris, 8M Urea, 500 mM 咪唑, pH8.0。 [6] IPTG 二、器材 摇床,离心机,层析柱(1 10 cm),蠕动泵 操作方法 一、氯霉素酰基转移酶重组蛋白的诱导 1. 接种含有重组氯霉素酰基转移酶蛋白表达载体的大肠杆菌BL21(DE3)菌株于5mL
蛋白质结构解析研究进展作业
《蛋白质结构解析研究进展》 一、蛋白质结构分类 人类对于进化的认识及蛋白质结构相似性比较的研究使蛋白质结构分类成为可能,而且近年来取得的研究进展表明,大部分蛋白质可以成功的分入到适当数目的家族中。目前国际上流行的蛋白质结构分类数据库基本上采取两种不同的思路,一种是数据库中储存所有结构两两比较的结果;第二种思路是致力于构建非常正式的分类体系。由于所有分类方法反映了各研究小组在探究这个重要领域的不同角度,所以这些方法是同等有效的。目前,被广泛应用的四种分类标准是:手工构造的层次分类数据库SCOP,全自动分类的MMDB和FSSP,和半手工半自动的CATH。 蛋白质结构自动分类问题可以被纳入机器学习的范畴,通过提取分析蛋白质结构的关键特征,构造算法来学习蕴含于大量已知结构和分类的数据中的专家经验知识,来实现对未知蛋白质结构的分类预测。目前,对蛋白质结构的不同层次分类,结果比较好的机器学习方法是:神经网络多层感知器、支持向量机和隐马尔可夫模型。支持向量机应用于分类问题最终归结于求解一个最优化问题。上世纪90 年代中期,隐马尔可夫模型与其他机器学习技术结合,高效地用于多重比对、数据挖掘和分类、结构分析和模式发现。多层感知器即误差反向传播神经网络,它是在各种人工神经网络模型中,在机器学习中应用最多且最成功的采用BP学习算法的分类器。 二、蛋白质结构的确定 蛋白质三维空间结构测定方法主要包括X射线晶体学分析、核磁共振波谱学技术和三维电镜重构,这三种方法都可以完整独立地在原子分辨水平上测定出蛋白质的三维空间结构。蛋白质数据库PDB中80%的蛋白质结构是由X射线衍射分析得到的,约15%的蛋白质结构是由核磁共振波谱学这种新的结构测定方法得到。 1、X射线晶体学
DNA和蛋白质的简单鉴别
一个简单的方法来区分蛋白质和DNA——一个普通化学的实验摘要:我们提出一个课堂实验,来补充被埃利斯等人在这个杂志提出的课堂活动。在埃利斯的活动中,学生从常见食物的细胞中提取高分子聚合物。在这个实验中,提取的聚合物是DNA还是蛋白质可以用做下面的三个易理解,便宜,而且容易的实验来鉴定。前两个测试——温度和酸介质效应——基于DNA的物理化学属性(可逆变性),第三个测试定性测定蛋白质(而非DNA)。这三种测试的结果提供证据来区分纯的的DNA分子和可能看起来像DNA的蛋白质。 艾利斯等人描述一个课堂活动,阐明了从动物和植物组织提取DNA的方法。在这个活动中,学生从常见的食物(牛肉、肝脏和洋葱)的细胞中用三个步骤提取DNA: (1)用机械粉碎的方法破坏细胞膜并添加缓冲洗涤溶液; (2)用离心机分离和除去细胞碎片, (3)通过添加冷乙醇沉淀DNA。 学生在最后一步获得一层DNA的高分子聚合物纤维。有什么证据能够证明这个聚合物纤维是DNA?我们如何能证明那些白色物质是DNA中而不使用分光光度计吗?学生们可能感觉提取的是蛋白质甚至认为沉淀是蛋白质,而不是DNA,所以我们如何区分蛋白质和DNA? 蛋白质是最丰富的生物大分子,存在于在所有细胞和细胞的所有部分。蛋白质是氨基酸的聚合物,每个氨基酸残基通过肽键这种特定的共价键和它旁边的氨基酸残基链接。蛋白质只能通过各种方法被分解(水解)成他们原来组成的氨基酸,所以,最早的对蛋白质研究集中在来源于他们的氨基酸。20种不同的氨基酸都在常见的蛋白质中发现,它们有一个羧基(-COOH)和一个氨基(-NH2)集团并且它们连接到同一个碳原子(α-碳)上。氨基酸的区别在于不同的侧链,或者说R基团不等的结构、尺寸、电荷。蛋白质有四级结构:一级结构、二级结构、三级结构和四级结构。所有共价键(主要是肽键和二硫键)对氨基酸残基的链接确定了多肽链的一级结构。最重要的决定二级结构的因素是指氨基酸残基的特定安排引起的循环结构 (螺旋和折叠)。三级结构说明了所有方面的多肽三维折叠方式。当一个蛋白质有两个或更多的多肽的子单元时,他们在空间的安排称为四级结构。了解DNA和蛋白质的主要理化特性,下面能完成这两种分子的区分。 我们提出一个课堂实验,来补充被埃利斯等人在这个杂志提出的课堂活动。这个实验在一个中学进行。活动30分钟即可进行完成。提取的聚合物是DNA还是蛋白质可以用做下面的三个易理解,便宜,而且容易的实验来鉴定。前两个测试——温度和酸介质效应——基于DNA的物理化学属性(可逆变性),第三个测试定性测定蛋白质(而非DNA)。这三种测试的结果提供证据来区分纯的的DNA分子和可能看起来像DNA 的蛋白质。 实验: 综述: 在这三个实验中,DNA的物理化学性质和蛋白质的不同。DNA样本在此前的一个实验中被“精制” (从草莓和香蕉中提取)。对于每个三个测试,获得的纤维绕在玻璃棒或木头棍(烧烤棒)上,像意大利面绕在叉子上一样,然后放在一个试管中。同样的
蛋白质组学及其主要技术
蛋白质组学及其主要技术 朱红1 周海涛2 (综述) 何春涤1, (审校) (1.中国医科大学附属第一医院皮肤科,辽宁沈阳110001; 2.北京大学深圳医院核医学 科,广东深圳518036) 【摘要】蛋白质组是指一种细胞、组织或有机体所表达的全部蛋白质。蛋白质组学是以蛋白质组为研究对象的新兴学科,近年来发展迅速,已成为后基因组时代的研究热点。目前,蛋白质组学研究技术主要包括:样品的制备和蛋白质的分离、蛋白质检测与图像分析、蛋白质鉴定及信息查询。本文就蛋白质组学概念及主要技术进行综述。 【关键词】蛋白质组,蛋白质组学 1蛋白质组学的概念 随着人类基因组测序计划的完成,人们对生命科学的研究重点由结构基因组转向功能基因组,1994年Wilkins和Williams首先提出蛋白质组一词[1],蛋白质组是指一种细胞、组织或有机体所表达的全部蛋白质。从基因到蛋白质存在转录水平、翻译水平及翻译后水平的调控,组织中mRNA丰度与蛋白质丰度不完全符合[2]。蛋白质复杂的翻译后修饰、蛋白质的亚细胞定位或迁移、蛋白质-蛋白质相互作用等也无法从DNA/mRNA水平来判断。因此,只有将功能基因组学与蛋白质组学相结合,才能精确阐明生命的生理及病理机制。 蛋白质组学是以蛋白质组为研究对象,对组织、细胞的整体蛋白进行检测,包括蛋白质表达水平、氨基酸序列、翻译后加工和蛋白质的相互作用,在蛋白质水平上了解细胞各项功能、各种生理、生化过程及疾病的病理过程等[3,4]。蛋白质组学有两种研究策略。一种是高通量研究技术,把生物体内所有的蛋白质作为对象进行研究,并建立蛋白质数据库,从大规模、系统性的角度来看待蛋白质组学,更符合蛋白质组学的本质。但是,由于剪切变异和翻译后修饰,蛋白质数量极其庞大,且表达随空间和时间不断变化,所以分析生物体内所有的蛋白质是一个耗时费力,难以实现的理想目标。另一种策略是研究不同状态或不同时期细胞或组织蛋白质组成的变化,主要目标是研究有差异蛋白质及其功能,如正常组织与肿瘤组织间的差异蛋白质,寻找肿瘤等疾病标记物并为其诊断治疗提供依据。 2蛋白质组学的常用技术 2.1样品的制备和蛋白质的分离技术 2.1.1样品的制备样品制备包括细胞裂解与蛋白质溶解,以及去除核酸等非蛋白质成分。 激光捕获显微切割(Laser-captured microdissection, LCM)[5]技术可大量获得足够用于蛋白质组学研究的单一细胞成分,避免其他蛋白成分对电泳结果的干扰。尤其是肿瘤的蛋白质组学研究常用LCM技术来获取单一的肿瘤细胞。 2.1.2蛋白质的分离技术 ①双向凝胶电泳(Two-dimensional electrophoresis, 2-DE):双向电泳方法于 l975年由O'Farrell[6]首先提出,根据蛋白质等电点和分子量的差异,连续进行成垂直方向的两次电泳将其分离。 第一向为等电聚焦(Isoelectric focusing,IEF)电泳,其基本原理是利用蛋白质分子的等电点不同进行蛋白质的分离。较早出现的IEF是载体两性电解质pH梯度,即在电场中通过两性缓冲离子建立pH梯度;20世纪80年代初建立起来的固相pH梯度(Immobilized pH gradients,IPG)IEF,是利用一系列具有弱酸或弱碱性质的丙烯酰胺衍生物形成pH梯度并参与丙烯酰胺的共价聚合,形成固定的、不随环境电场条件变化的pH梯度。IPG胶实验的重复
三种分析蛋白结构域的方法
三种分析蛋白结构域(Domains)的方法 1,SMART入门,蛋白结构和功能分析 SMART介绍 SMART (a Simple Modular Architecture Research Tool) allows the identification and annotation of genetically mobile domains and the analysis of domain architectures. More than 500 domain families found in signalling, extracellular and chromatin-associated proteins are detectable. These domains are extensively annotated with respect to phyletic distributions, functional class, tertiary structures and functionally important residues. Each domain found in a non-redundant protein database as well as search parameters and taxonomic information are stored in a relational database system. User interfaces to this database allow searches for proteins containing specific combinations of domains in defined taxa. For all the details, please refer to the publications on SMART. SMART(,可以说是蛋白结构预测和功能分析的工具集合。简单点说,就是集合了一些工具,可以预测蛋白的一些二级结构。如跨膜区(Transmembrane segments),复合螺旋区(coiled coil regions),信号肽(Signal peptides),蛋白结构域(PFAM domains)等。 SMART前该知道的 1,SMART有两种不同的模式:normal 或genomic 主要是用的数据库不一样。Normal SMART, 用的数据库 Swiss-Prot, SP-TrEMBL 和 stable Ensembl proteomes。Genomic SMART, 用全基因组序列。详细列表:,一些名词解释 进行时 可以直接用各个数据库蛋白的ID。如Uniprot/Ensembl??ID / Accession number (ACC)。或是直接蛋白序列。运行SMART也可选择signal peptides、PFAM domains等的预测,勾上就是。看下图 SMART结果 运行后的结果用图表表示。其实运行后的结果都有明确的解释。详细请看下面。