取代基效应 2
取代基效应

在21世纪,物理有机化学家将会在更广阔的范围内,在相关的前沿交叉领域中寻找新的学科生长点,运用自己在理论、方法、概念和思维方式方面的特长和优势,研究新问题,发现新规律,为有机化学乃至整个科学事业的发展作出贡献。
其中主要包括生命过程中的化学问题,分子聚集体化学中的结构/活性关系和反应规律,新分子和新材料的分子设计、合成和构效关系,计算化学和理论有机化学,自由基化学,有机光化学等领域。
内容提要§1-1 诱导效应一、共价键的极性与静态诱导效应 二、静态诱导效应的强度 三、静态诱导效应的强度比较 四、烷基的诱导效应 五、动态诱导效应 六、诱导效应对反应活性的影响 §1-2 共轭效应一、电子离域与共轭效应 二、静态共轭效应 三、动态共轭效应 四、共轭体系 五、共轭效应与反应性 §1-3 超共轭效应一、超共轭效应的特点和方向 二、超共轭效应的表现和作用 §1-4 场效应和空间效应 一、场效应 二、空间效应第一章 取代基效应(Substituent Effects)反应的本质: 有机化合物的反应本质是旧键的断裂,新键的生成,这直接或间接与共价键的极性,即共价键上电子云的分布有关。
例:C C C O1234取代基效应 分子中的某个原子或原子团对整个分子或分子中其它部分产生的影响(包括对共价键极性及整个分子物理性质和化学的影响)。
取代基效应的分类 取代基效应电子效应: 诱导效应 共轭效应 超共轭效应 场效应: 空间传递空间效应:空助效应 位阻效应 §1-1 诱导效应(Inductive effect) 一、共价键的极性与静态诱导效应C CH CH 21.定义CCCl ddd取代基的影响→分子链传递Cl d→电子云密度分布不均匀CCl d取代基性质→分子链传递方向δCH 3 CH CH 2 +→转移的结果存在于未发生反应的分子中——IS2.特点结构特征 单、双、叁键 传递方式沿价键链传递诱导效应的相对强度 取决于取代基中心原子电负性的大小;取代基的个数——加和性传递强度 距离越大,强度越弱 3.方向CZC H Z -I标准+I二、静态诱导效应的强度1.根据中心原子在元素周期表中的位置判断同周期 -IFOHNH 2CH 3OR 2NR 3 +IONR同主族 -I —F > —Cl > —Br > —IPR 2NR 3+IOS2.带正电荷取代基的-I 强,带负电荷取代基的 +I 强-I NR 2NR3NO 2+IOOR3. 中心原子相同 不饱和度越大,-I 效应越强 -ICCRCHCHROORN NRNR 2三、静态诱导效应的强度比较 相对次序比较 1.根据酸碱的强度比较测定取代乙酸的电离常数,诱导效应强度次序如下:-I 效应NO 2N(CH 3)3CNFClBrIOHOCH3C 6H 5CH CH 2H+I 效应C(CH 3)3HCH(CH 3)2CH 2CH 3CH 32.根据偶极矩比较测定甲烷一取代物和溴代烷的偶极矩,诱导效应强度次序如下: -I 效应NO2CNFCl BrIH+I 效应C(CH 3)3CH 2CH(CH 3)2CHCH 2CH 3CH 3CH 2(CH 2)2CH 33.根据1H NMR 化学位移比较测定X -CH3中甲基的 值,比较取代基的诱导效应强度。
高等有机化学 取代基效应自PPT课件
4、诱导效应强弱
诱导效应的相对强度:取决于取代基的中心原子 的电
负性 (Electronegativeties)
比氢原子相对电负性愈大,则-I效应愈强, 比氢原子相对电负性愈小,则+I效应愈强。
第12页/共94页
(1)与碳原子直接相连的原子
➢同周期的元素自左到右电负性增大,-I 增强:
第16页/共94页
例如:以乙酸为参考酸,具有X—CH2COOH通式, 由
于取代X基诱导效应pk的a 方向和X 强度不同,其pk酸a 性强弱不
同一NO2
1.68
一+N(CH3)3 1.83
一I —OH
3.12 3.83
一CN
2.46 —H
4.76
—F
2.66 —CH
4.88
—C1
2.86 一C(CH3)3 5.05
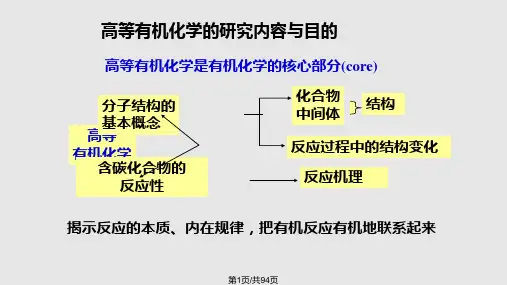
高等有机化学的研究内容与目的
高等有机化学是有机化学的核心部分(core)
分子结构的 基本概念 高等 有机化学 含碳化合物的
反应性
化合物 中间体
结构
反应过程中的结构变化
反应机理
揭示反应的本质、内在规律,把有机反应有机地联系起来
第1页/共94页
目录
第一章 取代基效应 第二章 有机反应活性中间体 第三章 饱和碳原子上的亲核取代反应 第四章 芳环上的取代反应 第五章 碳碳重键的加成反应 第六章 碳杂重键的加成反应 第七章 消除反应
但卤代烷的亲核取代反应活性却恰恰相反,实际其相 对活
性为: R—I>R—Br>R—C1 > C—F
原因:是动态诱导效应的影响。因为在同族元素中, 随原
子序数的增大电负性降低,其电子云受到核的约束也 第35页/共94页
第1章取代基效应.ppt

+B r
S 2 N
O H
C H 3 H CCC H 3 3 B r
H C 3 C H 3 C H 3
O H
C H 3 H CCC H 3 3 O H
S 1 N
3、 对反应速率的影响(快慢)
下列羰基化合物发生亲核加成的活性顺序为:
静态诱导效应 (Is) : 存在于未发生反应的分 子中的诱导效应(电负性决定) 诱导效应的传导 沿分子链迅速减弱 ,经过三 个原子之后,诱导效应已 很微弱 ,超过五个 原子便可忽略。 Ka×104 α-氯代丁酸 14.0 β-氯代丁酸 0.89 γ-氯代丁酸 0.26 丁酸 0.155
CO2H Cl Cl CO2H Cl CO2H CO2H
1.1.3关于烷基的诱导效应问题
C H 3 C H 3 C H 3 C H C H 3 C H 3 C H 3 C D
μ=0.132D μ=0.141D (已知氘比氢供电性略强)
C H 3 C H 2 C H 3
C H 3
C D 2
C D 2 C H 2 C D 2
μ=0.085D μ=0.095D
2、同周期元素的原子及其所形成的基团 在同一周期中,随着原子序数的增加, 元素的电负性增大,对电子的约束性增 大,因此极化性变小,故动态诱导效应 随原子序数的增加而降低。
Id:- CR3 > -NR2 > -OR > -F
1.1.5诱导效应对反应的影响
1、对反应方向的影响
H C 3 + H C l C l C 3 H 2 C l C H 3 C C 2 H H C C H C 3 3 C l H C C C H 3 3 H C l H 2 C l C H CC 3 2
取代基效应对锇(Ⅱ)羟基喹啉配合物结构和光谱性质的影响

摘要: 从理论上研究 了一系列 [ s C t ) O N) t O ( O) (f ( ](f a a=三氟 乙酸 ; =5氟一 0N - 羟基喹 啉 ( ) 羟 基 1,
喹啉 ( )和 2甲 基一 基 喹 啉 ( )配 合 物 的 结 构 和 光 谱 特 征.分 别 采 用 B L P L N 2 z 和 2 : 羟 3) 3 Y / A LD
根 据 Fa c - od n垂 直跃迁 原理 , 用含 时 密度 rn kC n o 利
[ s C )( ) O N ] O(O , ( ) 框架结构 的配合物 , 配 该 合物包含 3 个羰基配体 , 一个三氟 乙酸配体和一 个羟基喹啉配体. 它们 的最低 能吸收和发射波长 较短, 以 1 一 T 跃迁为 主, 且 T 1 金属 的参 与较少 , 配体起到了重要作 用 , 量子效率 只有 1%左 右. 0 为了能合成出具有较高磷光量子效率的配合物 , 实验工作者做了大量 的工作 , 但是还没有 比较成 熟的规律作为分子设计 的理论依据. 了研究这 为
21 0 1年 5月
文 章 编 号 :072 5 (0 1 0 -0 60 10 - 3 2 1 )50 2 -5 8
.
取 代 基 效 应 对 锇 (I羟 基 喹 啉 配 合 物 I ) 结 构 和 光 谱 性 质 的 影 响
张 建坡 , 金 丽
( 吉林化工学院 化学与制药工程学院’ , 吉林 吉林 12 2 ) 30 2
设计 中【 ]这些配合物最低能吸收和发射多以金 1. - 4
属 到 配 体 的 电 荷 转 移 ( C me lt lad ML T: t o i n a g
ca e r s r跃迁为主, hr a f ) g tn e 金属的参与对较低激发
第二章有机化学中的取代基效应

动态诱导效应和静态诱导效应在多数情况下是一致的,都属于极性 效应,但由于起因不同,有时导致的结果各异。例如:
C-X键的极性次序为: C-F > C-Cl > C-Br > C-I
例 如1, 3- 丁 二 烯 在 基 态 时 由 于 存 在 共 轭 效 应 , 表 现 体 系 能 量 降 低 , 电 子 云 分 布 发 生 变 化 , 键 长 趋 于 平 均 化 , 是 静 态 共 轭 效 应 的 体 现 。
当前您浏览到是第十八页,共三十四页。
动态共轭效应的体现
H2C
C H
卤代烷的亲核反应活性:R-I > R-Br > R-Cl > R-F
原因就是动态效应的影响。因为在同族元素中,随原子序数的增大电负性 降低,其电子云受到核的约束也相应减弱,所以极化性增大,反应活性增加 。
当前您浏览到是第十页,共三十四页。
二. 共轭效应 (Conjugative Effect)
1. 共轭体系与共轭效应
静态时:
(分子没有参加反应)
δC Cδ Cl
-I > +C
动态时:
(分子处于反应中〕
δ C δC Cl
+C > -I
当前您浏览到是第十七页,共三十四页。
动态共轭效应
与诱导效应相似,共轭效应也存在静态和动态的区别,
静态共轭效应是共轭体系内在的固有的性质,在基态时就存 在的。而动态共轭效应则是共轭体系在反应过程中,在外界 电场影响下所表现的瞬间的暂时效应。
取代基效应对二取代二苯基硝酮还原电位的影响

取代基效应对二取代二苯基硝酮还原电位的影响罗青青;曹朝暾;曹晨忠【摘要】合成了36种二取代二苯基硝酮XArCH =N(O)ArY(简称XPNY)化合物,研究了取代基效应对其还原电位(Ered)的影响,并系统对比了XPNY与XArCH=NArY(简称XBAY)和XArC(Me)=NArY(简称XPEAY)还原电位的差异.研究结果表明:XPNY的Ered与C=N键的13C NMR化学位移δc(C=N)没有线性关系;XPNY、XBAY和XPEAY三类化合物的Ered之间没有线性关系,表现出不同的变化规律;X基团的激发态取代基效应和间位基团位置指示变量对化合物XPEAY和XBAY的Ered都有贡献,而对化合物XPNY的Ered贡献都很小,可忽略;Y基团的激发态取代基效应对化合物XPNY的Ered有一定贡献,而对化合物XPEAY和XBAY 的Ered贡献很小,可忽略;XBAY和XPNY的母体有近似的还原电位,而XPEAY的母体其还原电位更低,一般而言,X-Y基团对相同时,XPEAY化合物更难被还原.【期刊名称】《物理化学学报》【年(卷),期】2016(032)007【总页数】8页(P1691-1698)【关键词】二苯基硝酮;取代基效应;化学位移;还原电位;激发态取代基参数;指示变量【作者】罗青青;曹朝暾;曹晨忠【作者单位】湖南科技大学化学化工学院,理论有机化学与功能分子教育部重点实验室,分子构效关系湖南省普通高校重点实验室,湖南湘潭411201;湖南科技大学化学化工学院,理论有机化学与功能分子教育部重点实验室,分子构效关系湖南省普通高校重点实验室,湖南湘潭411201;湖南科技大学化学化工学院,理论有机化学与功能分子教育部重点实验室,分子构效关系湖南省普通高校重点实验室,湖南湘潭411201【正文语种】中文【中图分类】O646含有C=N双键的共轭席夫碱是一类常见的,也是非常重要的化合物,在配位化学1-3、生物4-6、医药7-9、光电材料10,11以及谱学性能12-14等领域被广泛研究和应用.由于C=N双键易被还原成CH―NH饱和键,在有机合成上常常用于合成相应的胺类化合物,因而对它们的还原能力和还原电位也开展了一些研究15-18.通常席夫碱被还原首先发生在C=N极性键上,这已被实验证实17-20.Sauro和Workentin19对于对称的取代吖嗪还原电位研究,Zhu等20对单取代亚胺还原电位研究,Celik等21对单取代苯甲醛(苯乙酮)肟的还原半波电位研究,均表明C=N 键被还原的能力与取代基的电子效应(Hammett常数σ)22有密切关系.即取代基的吸电子效应利于C=N键的还原,供电子基阻碍C=N键的还原.然而,当将取代基的变化范围扩大到取代氮苄叉苯胺XArCH=NArY(N-(benzylidene)-anilines,简称XBAY),即C=N键两端的芳基上均连有取代基,且X、Y同时变化时,XBAY的还原电位(Ered)不能简单地采用取代基X、Y的Hammett 常数表达其变化规律,情况要比单取代基变化时复杂.曹晨忠等23对52种4,4′-二取代p-XBAY-p的还原电位研究表明,它们的Ered除了采用取代基Hammett常数外,还必须采用激发态取代基参数(σexCC)24一起表达其变化规律.该研究还表明,p-XBAY-p的Ered与C=N键的13C NMR化学位移(δC(C=N))不存在线性关系,即它们的Ered不是由C=N双键上碳原子的部分正电荷控制.同时曹晨忠等23还提出席夫碱得到一个电子后,表现出类似于激发态的电子分布,来解释取代基σexCC所起的作用.Wang等25对含间位取代基的XBAY还原电位的研究也表明,σexCC同样有重要作用.为了探讨激发态取代基参数σexCC对还原电位Ered的影响是否为普遍现象,Yuan 等26采用二取代氮-(苯亚乙基)苯胺XArC(Me)=NArY(N-(phenyl-ethylene)-anilines,简称XPEAY)的还原电位进行分析,发现其Ered受取代基影响与XBAY中有所不同,但同样需采用σexCC才能较好地定量XPEAY的Ered变化规律.他们还对其它几类有关化合物的Ered进行分析,得出:激发态取代基效应对席夫碱还原电位有重要影响.分析XBAY和XPEAY的分子结构发现,两者之间仅在于桥键不同,前者为CH=N,后者为C(Me)=N,即XPEAY相比XBAY仅在C=N双键的C端多了一个甲基Me(见图1),而它们的Ered变化规律却不同.由此,我们想到另一类化合物,二取代二苯基硝酮XArCH=N(O)ArY(N-phenyl-αphenylnitrones,简称XPNY,见图1),该类化合物的桥键比XBAY桥键的N端多了一个O原子,它们的Ered变化规律与XBAY和XPEAY有什么不同呢?或者说XBAY的CH=N键换成C(Me)=N、CH= N(O)键以后,其Ered受取代基X、Y的影响呈现出什么特点呢?这是一个令人感兴趣的理论问题,也是在有机合成中还原这三类化合物选择还原剂时需要考虑的问题.本文对XPNY的还原电位进行研究,并对上述三类化合物的还原电位变化规律进行对比,探讨席夫碱桥键的变化对还原电位的影响.2.1 材料准备目标化合物XPNY的合成参照文献27进行(如图2所示).所有化合物均用NMR进行表征(核磁数据和谱图见Supporting Information).2.2 数据准备2.2.1 支持电解质(四丁基胺六氟磷酸盐,(n-Bu)4NPF6)溶液的制备精密称取3.8745 g(0.01 mol)(n-Bu)4NPF6于100 mL容量瓶中,用无水无氧乙腈定容,得到0.1 mol.L-1的(n-Bu)4NPF6/CH3CN的支持电解质溶液.2.2.2 参比电极(Ag/AgNO3电极)的制作参比电极由电极玻璃套管,置于0.1 mol.L-1支持电解质溶液(包含0.01 mol.L-1AgNO3)中的Ag丝以及陶瓷芯三部分组接而成,置于上述组成的乙腈溶液中备用,使用前依次用蒸馏水、95%乙醇清洗,用冷风吹干.2.2.3 电化学测量所有化合物XPNY的还原电位Ered均在氮气保护下的无水无氧乙腈中使用CS300电化学工作站进行测定.电化学测量采取循环伏安法(CV),所用三电极的电极体系中,工作电极为玻碳电极,每次测量前均用氧化铝研料打磨,并依次用蒸馏水、95%乙醇、丙酮清洗,然后用冷风吹干;助电极为铂电极,每次测量前先将助电极依次用蒸馏水、95%乙醇、丙酮清洗,然后用冷风吹干;参比电极为自制的Ag/AgNO3电极,每次测量前先将参比电极依次用蒸馏水、95%乙醇冲洗,然后用冷风吹干;扫描速度为100 mV.s-1,用二茂铁校正后先测空白样,再测待测样.图3是一个典型的循环伏安图事例,测得的XPNY的还原电位值Ered列于表1.3.1 XPNY的Ered的相关分析Neuvonen等28研究表明,C=N双键上C所带的部分正电荷qC(C=N)与其C的NMR化学位移δC(C=N)有良好的正相关,于是我们首先想到XPNY的Ered是否与δC(C=N)有关,因此采用Ered对δC(C=N)作图,得到图4.由图4可知,两者并没有线性关系,说明XPNY的Ered不由C=N键上C原子的部分正电荷控制.然后,采用表1中XPNY的Ered数据,用相应的X、Y基团的Hammett常数和激发态取代基常数对Ered进行回归分析,优化后得到回归方程(1).(1)式中σ为取代基的Hammett电子效应常数,即σ=σF+σR.其中,σF为场/诱导效应,σR为共轭效应.例如,σF(X)和σF(Y)分别表示基团X和Y的场/诱导效应,σ(X)=σF(X)+σR(X).σexCC表示基团的激发态取代基参数,σexCC(X)和σexCC(Y)分别表示基团X和Y的激发态取代基参数.另外,R表示相关系数,S为标准偏差,F为Fisher检验值,n为回归方程中用到的数据点.方程(1)表明,X基团的诱导效应σF(X)和共轭效应σR(X)对Ered的影响同等重要,即两者可以合并在一起,但X的激发态取代基效应σexCC(X)作用很小,在方程中可以忽略;Y基团的共轭效应σR(Y)的作用可以忽略,它的诱导效应σF(Y)和激发态取代基效应σexCC(Y)的作用不能忽略.可见,在XPNY中,取代基X和Y对Ered的影响有差异.方程(1)的标准偏差为0.06 V,接近实验误差,相关系数为0.8450,可能原因是该系列中Ered的变化范围不太大(ΔEred=0.41 V).3.2 XPNY与XPEAY的Ered变化对比最近,Yuan等26对XPEAY的Ered进行分析,得到方程(2).(2)式中,Σσ表示取代基X和Y的Hammett参数之和,即Σσ=σ(X)+σ(Y).Δσ2是取代基X与Y之间的特殊交叉作用,即Im为区分对位和间位基团的指示变量,即间位基团Im=1,对位基团Im= 0.对比方程(2)和方程(1),可见C(Me)=N桥键换成CH=N(O)键后,取代基对Ered的影响差异较大.在XPEAY中,X、Y基团的σF、σR对Ered有相同作用,可以合并到Σσ一项之中,且X、Y的特殊交叉作用Δσ2也起作用.而在XPNY中X、Y基团的σF、σR对其Ered的作用不相同,不能合并到一起,且Y的σR可忽略,Δσ2的作用也不明显.激发态取代基效应σexCC的作用在XPEAY和XPNY中也有差异,前者的Ered主要受X的激发态取代基效应σexCC(X)的影响,后者的Ered则主要受Y的激发态取代基效应σexCC(Y)的影响.引起这些差异的原因可能是在XPEAY中Me 的供电子效应与C=N键π电子流向相同,在XPNY中O-的供电子效应与C=N键π电子流向相反(如图1分子结构中箭头所示).另外,在XPEAY中取代基处于间位时,还存在一种位置的特定作用,它由指示变量Im表示,使Ered产生-0.10 V的增量,而在XPNY中间位基团的位置特定作用不显著,定量相关方程中无需加入指示变量Im.3.3 XBAY、XPEAY和XPNY的Ered变化对比由于XBAY23,25、XPEAY26与本文XPNY三类化合物的Ered测试条件都一致,因而可以对这三类化合物的Ered进行比较分析.XBAY的还原电位测试数据见Supporting Information.为了直观比较XBAY、XPEAY和XPNY的Ered之间的变化情况,我们分别以XPEAY和XBAY的Ered的实验值对XPNY的Ered的实验值作图(例如p-ClPEAF-P vs p-ClPNF-p;p-ClBAF-P vs p-ClPNF-p),得图5.图5表明,含有相对应的X-Y基团对时, XPNY-XBAY以及XPNY-XPEAY之间的Ered没有线性关系,说明三者的Ered变化规律是不相同的,这意味着,三类化合物之间,不能以一类化合物的Ered变化趋势去预测另一类化合物的Ered变化趋势.对于XBAY,曹晨忠等23对52个4,4′-二取代p-XBAY-p的Ered进行定量相关,Wang 等25对49个3,4′/4,3′/3,3′-二取代XBAY的Ered进行定量相关,分别得到方程(3)和(4).Δσ是取代基X与Y的激发态取代基参数之间的特殊交叉作用,即方程(3)和(4)看上去好像表达不同的变化规律,但仔细分析,不论是4,4′-二取代XBAY还是3,4′/4,3′/ 3,3′-二取代XBAY,它们均共有同一母体结构氮苄叉苯胺(HBAH),这意味着,它们的Ered应该有同一变化规律.我们注意到,文献23得到的方程(3)采用的Ered未经二茂铁还原电位校正,文献25是经过校正的Ered.理论上同一母体结构的一系列化合物,其Ered应遵循相同变化规律.因而我们对文献23的Ered经过二茂铁还原电位校正(增加0.27 V),将文献23,25的样本放在一起构成一个数据集,进行回归分析(具体Ered数据见Supporting Information中的表S3),优化出统一方程(5).方程(5)表明,XBAY的Ered确有一致的变化规律.对比方程(5)的计算值和实验值发现,p-Cl-BACN-m和m-OMeBACN-m两个化合物的Ered的标准偏差较大,疑为离群值.试着去掉这两个Ered数据,再一次回归,得到方程(6).对比方程(6)和方程(5),可见方程(6)的结果明显优于方程(5).这表明方程(6)更接近真实地表达了XBAY的Ered的变化规律.于是,我们就可以通过方程(1)、(2)和(6)来比较XPNY、XPEAY和XBAY三类化合物Ered的变化规律.为了方便比较,将方程(1)、(2)和(6)列于表2,通过比较变量及变量之前的系数来了解三类化合物的Ered受取代基影响的异同.从表2中三类化合物Ered的定量方程分析,影响Ered的因素本质上可分为两种,一种是基态下中性分子从外界(电极)获得电子的能力,另一种是分子自由基负离子M.-(分子从电极中得到一个电子形成自由基负离子)的稳定性程度.前者由基态下取代基电子效应常数(Hammett常数σ)表示,后者(类似于激发态23)由激发态取代基常数σexCC表示.三类化合物的Ered受σ参数的影响总体上一致,即取代基X、Y的吸电子效应(σ为正)使分子易于得到电子而容易被还原(Ered升高).因而方程(1)、(2)和(6)分别表达了基态下取代基电子效应参数σ和激发态取代基参数σexCC对XPNY、XPEAY和XBAY的还原电位的影响.由于XPNY、XPEAY和XBAY三类化合物的桥基分别为CH=N(O)、C(Me)=N和CH=N,母体分子骨架的微小变化导致它们各自的Ered受X、Y基团的具体影响存在差异.以XBAY系列为参考基准,在XBAY中,X、Y的Hammett常数σ对Ered 均有影响,σ(X)和σ(Y)前面的系数为正值,σ(X)和σ(Y)越正,越易得到电子,说明基态下基团X、Y的吸电子效应使Ered增加,使XBAY分子易于还原,而供电子效应使Ered降低(难于还原),且X基团的影响大于Y基团的影响.X基团的激发态效应对Ered有影响,而Y基团的激发态效应对Ered影响很小,可忽略.当σexCC为正值时减小Ered,为负值时增加Ered.通常,基团的激发态取代基参数σexCC为负值,在方程(6)中,σexCC(X)前面的系数为负值,说明X基团的激发态取代基电子效应使XBAY分子得到电子形成的分子自由基负离子[XBAY].-比较稳定,利于分子还原.另外,基团处于间位的位置对Ered有特定影响,使Ered有-0.20 V的变化.当XBAY中桥键CH=N换成C(Me)=N后,即在XPEAY中,X、Y的Hammett常数σ对Ered有相同影响,可用Σσ表达(方程(2)中Σσ前面的系数为正,表明基态下X、Y的吸电子效应使分子易于还原,并且还存在X-Y基团之间的特殊交叉相互作用(Δσ2).σexCC(X)前面的系数为正值,说明X基团的激发态取代基电子效应使XPEAY 分子难于还原, XPEAY与XBAY类似,也存在基团间位位置的特定影响,它使Ered 产生-0.10 V的增量.当XBAY中桥键CH=N换成CH=N(O)后,即在XPNY中,X基团的Hammett常数σF(X)和σR(X)有相同影响,可以合并成σ(X)一项,而Y基团只有诱导效应σF(Y)起重要作用,其共轭效应σR(Y)的作用可忽略.且在方程(1)中,σ(X)和σF(Y)前面的系数均为正值,说明基态下基团X、Y的吸电子效应使Ered升高,即分子易于还原.但在XPNY中,X基团的激发态取代基效应σexCC(X)作用很小,可忽略.相反,基团Y的激发态取代基效应σexCC(Y)有重要作用,并且前面的系数为正值,说明Y基团的激发态取代基电子效应使XPNY分子还原形成的自由基负离子[XPNY].-不稳定,不利于还原.我们还注意到,在XPNY化合物中,间位基团没有表现出位置的特定影响,方程中无须使用指示变量Im.就方程(1)、(2)和(6)的截距而言,(1)、(6)的截距几乎相等,而(2)的截距相差-0.30左右.以上说明XBAY和XPNY 的母体有近似的还原电位,而XPEAY母体的还原电位更低,更难于被还原.图6是具有相同X-Y基团对的XPNY、XBAY和XPEAY的Ered大小对比,可见,在X-Y基团对相同时,XPEAY具有更小的Ered值(更负),即它比相应的XBAY和XPNY更难还原.通过对XPNY的还原电位的系统分析及其与XBAY和XPEAY还原电位对比分析,可以得出如下结论.(1)XPNY的Ered与C=N键的δC(C=N)不存在线性关系.即Ered不是受C=N键的C原子上的部分正电荷控制.(2)XPNY、XBAY和XPEAY三类化合物Ered之间没有线性关系,表现出不同的变化规律,在预测各自的Ered时不能相互类推其变化规律.(3)X的激发态取代基参数σexCC(X)和间位的位置指示变量Im对化合物XPEAY 和XBAY的还原电位Ered都有贡献,而对化合物XPNY的还原电位Ered的贡献都很小,可忽略.(4)Y的激发态取代基参数σexCC(Y)对化合物XPNY有一定贡献,而对化合物XPEAY和XBAY的贡献很小,可忽略.(5)XBAY和XPNY的母体有近似的还原电位,而XPEAY母体的还原电位更低,这意味着,在X-Y基团对相同时,XPEAY更难被还原.本文的研究有利于理解芳基席夫碱类化合物的分子结构对其还原电位的影响,也可以通过估算该类化合物的还原电位,在有机合成中还原C= N双键形成CH―NH键时,为还原剂的选择提供理论参考.Supporting Information:available free of charge via the internet at.References(1)Keypour,H.;Shooshtari,A.;Rezaeivala,M.;Kup,F.O.; Rudbari,H.A.Polyhedron 2015,97,75.doi:10.1016/j. poly.2015.02.029(2)Belal,A.A.M.;El-Deen,I.M.;Farid,N.Y.;Zakaria,R.;Refat,M.S.Spectrochim.Acta,Part A 2015,149,771.doi:10.1016/j. saa.2015.05.005 (3)Kal,S.;Filatov,A.S.;Dinolfo,P.H.Inorg.Chem.2014,53,7137.doi:10.1021/ic500 121f(4)Misra,S.;Pandeya,K.B.;Tiwari,A.K.;Ali,A.Z.;Saradamani,T.;Agawane,S.B.;Madhusudana,K.Med.Chem.Res.2011,20,1431.doi:10.1007/s00044-010-9377-3(5)Rahim,F.;Ullah,K.;Ullah,H.;Wadood,A.;Taha,M.;Rehman,A.U.;Uddin,I.;Ashra f,M.;Shaukat,A.;Rehman,W.;Hussain,S.;Khan,K.M.Bioorg.Chem.2015,58,81.doi:10.1016/j.bioorg.2014.12.001(6)Ren,S.J.;Wang,R.B.;Komatsu,K.;Krause,P.B.;Zyrianov,Y.;McKenna,C.E.;Csipke,C.;Tokes,Z.A.;Lien,E.J.J.Med.Chem.2002,45(2),410.doi:10.1021/jm010252q(7)Shahabadi,N.;Kashanian,S.;Darabi,F.Eur.J.Med.Chem.2010,45,4239.doi:10. 1016/j.ejmech.2010.06.020(8)Squarcialupi,L.;Colotta,V.;Catarzi,D.;Varano,F.;Filacchioni,G.;Varani,K.;Corc iulo,C.;Vincenzi,F.;Borea,P.A.;Ghelardini,C.;Mannelli,L.D.C.;Ciancetta,A.;Moro,S.J.Med.Chem.2013,56,2256.doi:10.1021/jm400068e(9)Sinha,D.;Tiwari,A.K.;Singh,S.;Shukla,G.;Mishra,P.;Chandra,H.;Mishra,A.K.Eur.J.Med.Chem.2008,43,160.doi:10.1016/j.ejmech.2007.03.022(10)Rajendran,A.;Tsuchiya,T.;Hirata,S.;Iordanov,T.D.J.Phys. Chem.A 2012,116,12153.doi:10.1021/jp3084315(11)Lei,S.D.;Wen,F.F.;Li,B.;Wang,Q.Z.;Huang,Y.H.;Gong,Y.J.;He,Y.M.;Dong,P.;B ellah,J.;George,A.;Ge,L.H.;Lou,J.;Halas,N.J.;Vajtai,R.;Ajayan,P.M.Nano Lett.2015,15,259.doi:10.1021/nl503505f(12)Cao,C.T.;Wei,B.Y.;Cao,C.Z.Acta Phys.-Chim.Sin.2015,31(2),204.[曹朝暾,魏佰影,曹晨忠.物理化学学报,2015,31(2),204.]doi:10.3866/PKU.WHXB201412191(13)Cao,C.Z.;Fang,Z.J.Spectrochim.Acta,Part A 2013,111,62.doi:10.1016/j.saa.2013.03.082(14)Cao,C.Z.;Lu,B.T.;Chen,.Chem.2011,24,335.doi:10.1002/poc.1760(15)Li,B.Y.;Yao,Y.M.;Wang,Y.R.;Zhang,Y.;Shen,Q.Inorg.mun.2008,11,349.doi:10.1016/j. inoche.2007.12.035(16)Szostak,M.;Spain,M.;Procter,.Chem.2014,79,2522.doi:10.1021/jo4028243(17)Rao,C.N.;Hoz,S.J.Am.Chem.Soc.2011,133,14795. doi:10.1021/ja205885q(18)Rao,C.N.;Hoz,.Chem.2011,76,9438.doi:10.1021/ jo2018153(19)Sauro,V.A.;Workentin,.Chem.2001,66(3),831.doi:10.1021/jo0056287(20)Zhu,X.Q.;Liu,Q.Y.;Chen,Q.;Mei,.Chem.2010,75(3),789.doi:10.1021/jo902332n(21)Celik,H.;Ekmekci,G.;Ludvík,J.;Picha,J.;Zuman,P.J.Phys. Chem.B2006,110(13),6785.doi:10.1021/jp056808t(22)Hansch,C.;Leo,A.;Taft,R.W.Chem.Rev.1991,91(2),165.doi:10.1021/cr00002a004(23)Cao,C.Z.;Bi,Y.K.;Cao,.Chem.2015,35, 1302.[曹晨忠,毕亚坤,曹朝暾.有机化学,2015,35,1302.] doi:10.6023/cjoc201411037(24)Cao,C.Z.;Chen,G.F.;Yin,.Chem.2008,21,808.doi:10.1002/poc.1387(25)Wang,L.Y.;Cao,C.T.;Cao,C.Z.Chin.J.Chem.Phys.2016,29,260.doi:10.1063/1674-0068/29/cjcp1508173(26)Yuan,H.;Cao,C.T.;Cao,Z.Z.;Chen,C.N.;Cao,C.Z..Chem.2016,29,145.doi:10.1002/poc.3511(27)Liu,S.J.;Wang,Y.H.;Jiang,J.Y.;Jin,Z.L.Green Chem.2009,11,1397.doi:10.1039/b906283a(28)Neuvonen,H.;Neuvonen,K.;Fulop,.Chem.2006,71, 3141.doi:10.1021/jo0600508。
第二章 取代基效应2
O C
< pKa
OH
当t-Butyl 在邻位时, 把羧基挤出了与苯环所在 平面,烃基的+C效应消失。
O N O O H
OH
OH O29.89
7.15
0.38
OH H3C CH3 H3C NO2
OH
CH3 NO2
pKa = 7.16
pKa = 8.24
3. 对反应活性的影响
2,6-二甲基吡啶 几乎不与R3B作用
R R R N
R' B R' R'
CH3 N CH3
在相互接近时基团位阻 导致相互排斥作用 F-张力(Face - Strain)
R C X R 109° 28' R
120°
R C + X R
R
SN1反应 形成正碳离子的一步 键角的变化缓解了 基团的拥挤程度 离去基团背后的张力 B-张力 (Back Strain)
H H COOH
酸性 较弱
酸性 较强
五. 空间效应 Space effect
分子内或分子间 不同取代基相互接近时 由于取代基的体积大小、形状不同 相互接触而引起的 物理的相互作用
空间效应的作用
1. 化合物(构象)的稳定性
H
CH3 H H CH3
2. 化合物的酸碱性
O C OH CH3 C CH3 CH3 pKa
中线以上 为反键轨道 α-xβ
0
中线表示非键轨道
中线以下 为成键轨道 α+xβ
不同电子体系分子轨道能级
3原子 α-β α+2β
4原子 α-2β α α+2β
5原子 α-1.60β α+0.62β α+2β
第二章 取代基效应
二、共轭效应与诱导效应的区别
(1)共轭效应起因于电子的离域,而不仅是极性 或极化的效应。 (2)共轭效应只存在于共轭体系中,不象诱导效 应那样存在于一切键中。 (3)诱导效应是由于键的极性或极化性沿σ键传 导,而共轭效应则是通过π电子的转移沿共轭链传递, 是靠电子离域传递;共轭效应的传导可以一直沿着 共轭键传递而不会明显削弱,不象诱导效应削弱得那 么快,取代基相对距离的影响不明显,而且共轭链 愈长,通常电子离域愈充分,体系能量愈低愈稳定, 键长平均化的趋势也愈大。
2、 动态诱导效应强度的比较
动态诱导效应是一种暂时的效应,不一定反映在分 子的物理性质上,不能由偶极矩等物理性质的测定来比 较强弱次序。比较科学、可靠的方法是根据元素在周期 表中所在的位置来进行比较。 ( 1 )在同一族元素,由上到下原子序数增加,电负 性减小,电子受核的约束减小,电子的活动性、可极化 性增加,动态诱导效应增强。如: Id : —I > —Br > —Cl > —F —TeR > —SeR > —SR > —OR
4、 静态诱导效应的强度
诱导效应的强度
主要取决于有关原子或基团的电负性, 与氢原子相比,电负性越大-I效应越强, 电负性越小则+I越强。 -I效应:—F > —OH > —NH2 > —CH3;
OR2 > NR3
+
+
+I效应:
NR >
-
O
-
中心原子带有正电荷的比不带正电荷的 同类基团的吸电诱导效应强,而中心原子带 有负电荷的比同类不带负电荷的基团供电诱 导效应要强。 + NR2 [例如] -I效应: NR3 > +I 效应: —O- > —OR 如果中心原子相同而不饱和程度不同,通常 随着不饱和程度的增大,吸电的诱导效应增 强。 [例如] =O > —OR ≡N > =NR > —NR2
取代基效应
取代基效应: 分子中的某个原子或原子团对整个分子或分子 中其它部分产生的影响。
电子效应
诱导效应 (σ,π) 共轭效应 ( π-π, p-π) 超共轭效应(σ- π, σ-p)
取代基效应
Hale Waihona Puke 场效应空间效应 (位阻效应)
电子效应 (Electronic effect): 由于取代基的作用而导致的共有电子对沿共价 键转移的结果。
- + + +
吸电子诱导效应:-I
+ - - 给电子诱导效应:+I
诱导效应的传导方式: 诱导效应的传导是以静电诱导的方式沿着单键或重 键传导的,只涉及到电子云密度分布的改变,引起 键的极性改变,一般不引起整个分子的电荷转移、 价态的变化。 结构特征: 单、双、叁键 传递方式:σ、π键 传递强度:与距离相关。距离越 大,强度越弱。
+I效应:
常见吸电子基团(-I): -+NR3,-+SR2,-+NH3,-NO2,-SO2R,-CN, -COOH,-OAr,-CO2R , -COR,-OH, -C≡CR,-Ar,-CH=CR2等。 -I:-NO2 >-N+(CH3)3 >-CN >-F >-Cl >-Br >-I >-OH 常见给电子基团:烷基,烷氧基,O-,-COO(+I):-C(CH3)3 > -CH2CH3 > -CH3
C. 相同的原子:不饱和度越大,-I 效应越强。 D. 带正电荷的取代基的-I效应强, 带负电荷的取代基的 + I效应强。 E. 甲基比氢原子易极化,甲基属于+I效应的基团。 +I: -C(CH3)3 > -CH(CH3)2 > -CH2CH3 >CH3
第一章取代基效应
2.16 0.23
上页 下页 返回 退出
(2)与周期表的关系(电负性)
同族: –F> –Cl> –Br> –I, –OR> –SR> –SeR 同周期: –F> –OR> –NR2 (3)不饱和度:
C CR
(4)带电荷: - –O-(+I) 同周期: –NR> + –NR3> –NR2 ( –I) 7.烷基的诱导效应问题 (1)实验数据 ① 酸性
超共轭效应 (σ- π,σ- p)
空间效应 (位阻效应) 物理的相互作用 习题
9
上页 下页 返回 退出
第一节 诱导效应 (Inductive effect)
C
δ
δ
C
δ
δ
Cl Cl Cl
δ
取代基的影响
分子链
C C C
δ
传递
电子云密度
取代基性质
C
δ
分布不均匀 方向
转移的结果
一. 静态诱导效应
首先观察下列两组数据
多数情况下,动态与静态的作用一致
15
上页 下页 返回 退出
2.动态诱导效应的相对强度:
同族: –F < –Cl < –Br < –I, –OR < –SR < –SeR 同周期: –F < –OR < –NR2 带电荷: O > OR > OR2
NR2 > NR3
三. 诱导效应对反应性能的影响
1.对反应方向的影响
-C :不饱和官能团
同周期 同族 (2)带电荷
C NH > C CH2
C O >
C S
-C : +C :
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
▪ 1H-NMR
▪ CH3-F CH3-Cl CH3-Br CH3-I CH3-H
▪ δ 4.3 3.2
2.2 2.2 0.23
▪ CHCl3 CHCl2 CH3Cl ▪ δ 7.3 5.3 3.1
▪ 诱导效应具有加合性。
第9页/共60页
a
ab CH3CH2Br
b
吸收强度
6
5
4
3
2
化学位移
第10页/共60页
1
0
3.通过测定偶极矩确定
化合物(偶极矩u/10-30(c.m) :
CH4 CH3NO2 CH3CHO CH3Cl CH3OH
0 11.80
8.97
6.10
5.64
-I 基团:
-NO2 > -CHO > -Cl > -OH +I基团:
(CH3)3C- > CH3CH2(CH3)CH- > CH3(CH2)2CH2> -CH3
第15页/共60页
烷基的电负性
H -CH3 -CH2CH3 -CH(CH3)2 -C(CH3)3
电负性: 2.1 2.2
2.21
2.24
2.26
第16页/共60页
2.当烷基连在不饱和碳原子或带正电荷的氮或氧 原子上,表现为给电子基。
Eg. CH3→CH=CH2 ; CH3→C≡CH
CH3
;
在多数情况下,烷基可看作给电子基。 (有人认为这是超共轭效应引起的)
第7页/共60页
2.通过分析NMR化学位移确定
化学位移δ大,电子云密度低,取代基-I强。 (* 电子密度大,屏蔽大,需要的外加磁场H大,即 小;
电子密度小,屏蔽小,需要的外加磁场H小,即 大。)
吸收强度
低场
高场 磁场强度H(↗)吸收强度
低场
高场 磁场强度H(↗)
第8页/共60页
卤代烃的1H-NMR δ值
吸电子诱导效应:-I;给电子诱导效应:+I
第3页/共60页
pKa 4.76
2.86
第4页/共60页
诱导效应( inductive effect)
δ +I
Y
标准
δ
δ
CR3 H
CR3 X
-I δ
CR3
随着距离的增大效应迅速减弱。
第5页/共60页
特点: 1. 起源于电负性;
2.是一种静电作用,在键链中传递 只涉及到电子云密度分布的改变,即主要 是键的极性的改变,且极性变化一般是单 一方向的;
取代基效应 场效应 (空间传递)
空间效应 (位阻效应)
结构1.诱导效应(Inductive effect)
一、诱导效应的涵义和特征
在有机化合物的分子中,由于电负性不同的 取代基的影响沿着键链(单链或重链)传递,致 使分子中电子云密度,按取代基相对氢的电负性 强弱所决定的方向而偏移的效应,叫诱导效应 (I)。 方向:
极化度大小取决于外界电场和分子体系电子云 流动性的大小。
第18页/共60页
(1)同族元素的原子及其原子团
Id : -I >-Br>-Cl>-F 带正电的原子团束
Id :
-O- >-OR>-O +R2
缚电子云的能力大; 带负电荷反之。
- NR2 >- N+R3
(2)同周期元素的原子及其原子团 电负性越大, 极化度越小。
第11页/共60页
(二)影响取代基诱导效应相对强度的有 关因素: 1.周期律
同周期中,-I效应自左至右增加。 Eg.
-I: -CR3 < -NR2 < -OR < -F 在同一族中,-I效应自上而下降低。 Eg. -I: -F > -Cl > -Br > -I;
-OR > -SR
第12页/共60页
第17页/共60页
四、静态诱导效应和动态诱导效应
静态诱导效应(IS):未发生化学反应时分子 中电子云密度分布状况的变化,基态分子的永久 极性。
动态诱导效应(Id):外来试剂的极性核心接近 反应分子时,尤其是试剂对分子的起始进攻而导 致过渡态的形成时,会引起分子中电子云密度分 布状态的变化,暂时的极化效应。这种由于外界 电场引起电子云分布状态的改变称之为动态诱导, 即可极化性。
第14页/共60页
三、烷基的诱导效应
目前认为烷基的诱导效应方向取决于与什么样 的原子(团)相连。如果与电负性比它大的原子 (团)相连,则表现出+I;如果与电负性比它 小的原子(团)相连,则表现出-I。 1.当烷基连在SP3杂化的碳原子上或中性的原子上, 表现为吸电子基。 脂肪醇在气相中的相对酸性顺序(与溶液中相反): (CH3)3COH > (CH3)2CHOH > CH3CH2OH > CH3OH > HOH
3.传递有一定限度,经过三个碳原 子以后,已极微弱(短程)
第6页/共60页
二.诱导效应的相对强度
(一)确定方法 1.通过测定取代酸、碱离解常数确定
①-I:-NO2 >-N(CH3)2 >-CN >-F >-Cl >-Br >-I >-OH
②+I: -C(CH3)3 > -CH2CH3 > -CH3
电离后,变成-COO-,对另一羧基而言为给电子基。
第13页/共60页
3.饱和程度 与碳原子相连的原子,若是同种原子
但饱和程度不同,则不饱和程度高的吸电 子能力强。
–I效应: -C≡CR > -CR=CR2 > -CR2-CR3 =O > -OR ≡N > =NR > -NR2
(电负性:CSP > CSP2 > CSP3)
教学目的和要求
▪ 1、掌握电子效应(诱导、共轭、超共轭) 的基本概念(定义、方向、强弱、传导)
▪ 2、掌握电子效应和空间效应对性能的影响 ▪ 3、了解场效应的概念及其对化合物性能的
影响 ▪ 4、学会用电子效应和空间效应解释和预测
反应现象。
课时安排:4h
第1页/共60页
电子效应
诱导效应 (σ, π) 共轭效应 ( π-π, p-π) 超共轭效应 (σ- π,σ- p)
2.电荷 带正电荷的基团比同类型的不带电荷的基团吸电子能
力强得多;
而带负电荷的基团比同类型的不带电荷的基团给电子 能力强得多。
Eg. –I效应:-NR3+ > -NR2 +I效应:-O- > OR
同类基团电荷的变化可改变诱导效应的方向或强度。
COOH
Eg.
CH2 COOH
,
-COOH为另一-COOH的吸电子基;
Id : -CR3 >-NR2>-OR>-F
第19页/共60页
在化学反应过程中,动态诱导起主导作用。 如RX发生SN反应的活性次序为:
RI>RBr>RCl
π键比σ活泼
第20页/共60页
五、诱导效应对反应性能的影响