制药企业全套检验记录()
药材检验原始记录样本
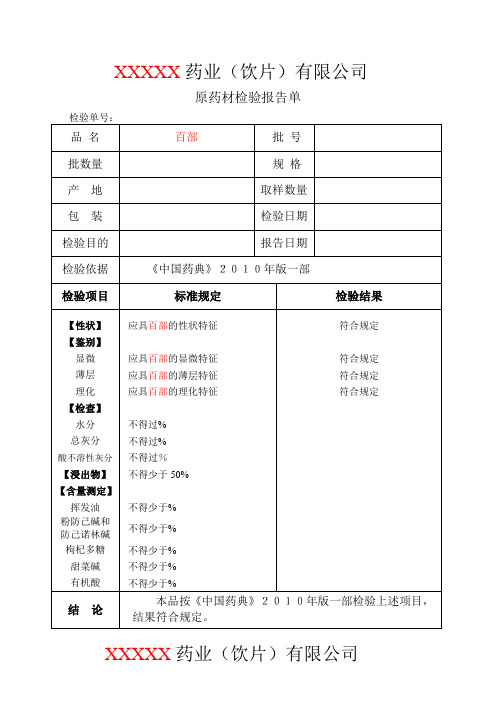
XXXXX药业(饮片)有限公司原药材检验报告单XXXXX药业(饮片)有限公司原药材检验记录【性状】结果:【鉴别】(1)显微鉴别横截面:结果:粉末:结果:(2)薄层鉴别供试品溶液的制备:取粉末1g,加乙醇15ml,加热回流1小时,放冷,滤过,滤液蒸干,残渣加乙醇5ml使溶解。
对照药材、对照品溶液配制:取菊花对照药材1g,同法制成对照药材溶液。
再取绿原酸对照品,加乙醇制成每1ml含的溶液。
温度:(℃)相对湿度:(%)展开剂:三氯甲烷-丙酮-甲醇-5%浓氨试液(6:1:1:)薄层板:硅胶G显色剂:稀碘化铋钾试液灯光:白光、紫外光灯(365nm)展距:(cm)供试品色谱中,在与对照药材色谱相对应的位置上,显相同颜色的荧光斑点。
S1为对照药材(对照品为中检所提供编号为)S2为对照品(对照品为中检所提供编号为) T为样品结果:【检查】杂质不得过 XX % (附录IX A)杂质称重: g杂质计算结果为: % (标准规定不得过 XX %)结果:膨胀度应不低于(附录IX O)温度:(℃)相对湿度:(%)电子天平型号:CP214 溶剂:水样品编号 1# 2# 3#干燥品称重: g g g第一次样品膨胀后体积: ml ml ml第二次样品膨胀后体积: ml ml ml(两次差异不超过)膨胀度计算结果为:(标准规定不低于)结果:水分不得过% (附录Ⅸ H 第一法)。
温度:(℃)相对湿度:(%)烘箱型号:DHG-91012SA型电子天平型号:CP214样品编号 1# 2#第一次称量瓶干燥(105℃ 3h) (g)(g)第二次称量瓶恒重(105℃ 1h) (g)(g)样品称重(g)(g)第一次称量瓶+样品干燥(105℃ 5h) (g)(g)第二次称量瓶+样品恒重(105℃ 1h) (g)(g)水分计算结果为:(%)(标准规定不得过%)结果:总灰分不得过%(附录Ⅸ K)温度:(℃)相对湿度:(%)马福炉型号:电子天平型号:CP214样品编号 1# 2#第一次坩锅称重(600℃ 3h) (g)(g)第二次坩锅恒重(600℃(g)(g)样品称重(g)(g)第一次坩锅+残渣称重(600℃ 3h) (g)(g)第二次坩锅+残渣恒重(600℃(g)(g)总灰分计算结果为:(%)(标准规定不得过%)结果:酸不溶性灰分不得过%(附录Ⅸ K)。
最新中药生产企业GMP中药提取现场检查记录
5、稠膏相对密度、性状;
6、稠膏收率%;
醇沉
1、场地、设备、容器具清洁;
2、物料名称、数量(称量核对);
3、物料标签;操作间、设备状态标识;
4、乙醇含量%;
5、沉淀时间h、效果;
回收乙醇并浓缩
1、生产场地、设备、容器具清洁,使用前必要的消毒;
2、回收乙醇浓度%;
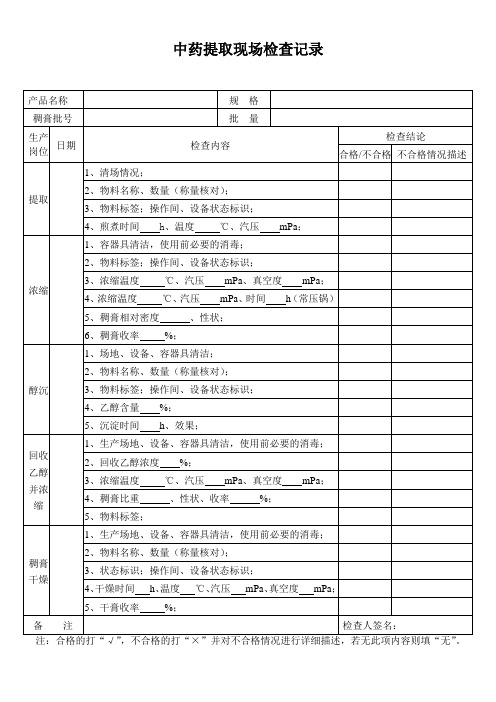
中药提取现场检查记录
产品名称
规格
稠膏批号
批量
பைடு நூலகம்生产
岗位
日期
检查内容
检查结论
合格/不合格
不合格情况描述
提取
1、清场情况;
2、物料名称、数量(称量核对);
3、物料标签;操作间、设备状态标识;
4、煎煮时间h、温度℃、汽压mPa;
浓缩
1、容器具清洁,使用前必要的消毒;
2、物料标签;操作间、设备状态标识;
3、浓缩温度℃、汽压mPa、真空度mPa;
3、浓缩温度℃、汽压mPa、真空度mPa;
4、稠膏比重、性状、收率%;
5、物料标签;
稠膏
干燥
1、生产场地、设备、容器具清洁,使用前必要的消毒;
2、物料名称、数量(称量核对);
3、状态标识;操作间、设备状态标识;
4、干燥时间h、温度℃、汽压mPa、真空度mPa;
5、干膏收率%;
备注
检查人签名:
注:合格的打“√”,不合格的打“×”并对不合格情况进行详细描述,若无此项内容则填“无”。
GMP药品检验记录大全

页码
备注
包装材料检验记录一览表
序号
记录名称
1 口服固体药用高密度聚乙烯瓶、盖检验记录
2 口服液体药用高密度聚乙烯瓶、盖检验记录
3 口服液体药用聚丙烯量杯检验记录
4 药品包装用铝箔检验记录
5 聚氯乙烯固体药用硬片检验记录
6
聚氯乙烯|低密度聚乙烯药品包装用复合硬片 检验记录
7
聚酯|铝|聚乙烯药品包装用复合膜、袋检验记 录
QS3006 A01 R01 V01
QS3007 A01 R01 V01
QS3008 A01 R01 V01 QS3009 A01 R01 V01 QS3010 A01 R01 V01 QS3011 A01 R01 V01 QS3012 A01 R01 V01 QS3013 A01 R01 V01 QS3014 A01 R01 V01 QS3015 A01 R01 V01 QS3016 A01 R01 V01 QS3017 A01 R01 V01 QS3018 A01 R01 V01 QS3019 A01 R01 V01 QS3020 A01 R01 V01 QS3021 A01 R01 V01
8 药用手指套检验记录
9 低硼硅璃安瓿检验记录
10 注射用冷冻干燥用卤化丁基橡胶塞检验记录
11 低硼硅玻璃管制注射剂瓶检验记录
12 抗生素用铝塑组合盖检验记录
13 标签检验记录
14 说明书检验记录
15 小盒检验记录
16 中盒检验记录
17 大箱检验记录
18 吸塑、托盘检验记录
19 内卡、遮光套检验记录
药品生产企业检查记录表
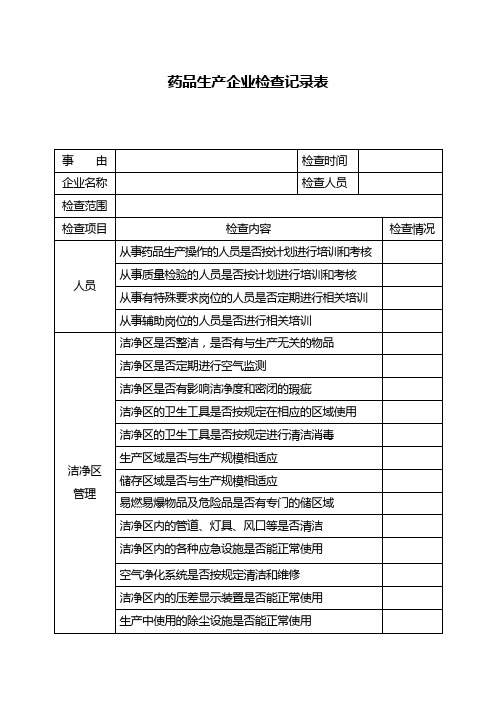
质量管理部门是否按规定对生产用水进行检验
质量管理部门是否按规定取样和留样
留样室的管理是否符合要求
质量管理部门是否按规定对标准品、滴定液进行标定
质量管理部门是否按规定对检验设备、量具进行校正
标准品、滴定液等是否按规定进行管理
检验用试剂、培养基等是否按规定条件储藏
空气净化系统是否按规定清洁和维修
洁净区内的压差显示装置是否能正常使用
生产中使用的除尘设施是否能正常使用
生产中使用的器具是否按规定清洗和消毒
有特殊要求的物品是否使用专用的器具
仪器设备管理
生产设备是否有明显的状态标志
生产和检验用仪器、量具、衡器等是否定期校验
有毒物品的衡器是否专用
生产设备是否按规定进行维修保养
质量管理部门是否定期对物料供应商进行评估
销售管理
产品销售是否有能追查到每批产品售情况的记录
产品退货和回收是否有能反映处理过程的记录
因质量原因退货和收回的产品,是否有详细的处理记录
是否建立药品不良反应监测制度
对有关药品质量和投诉和药品不很反应,是否有进行调查处理并有记录
纯化水系统是否按规定清洁消毒
物料管理
原辅料是否从指定的供应商购进有明显的物料标志及状态标志
原辅料是否经过检验合格后方投入使用
原辅料是否按规定条件储藏,并分别存放
原辅料发放是否有详细记录
进口原料药、中药材、中药饮片是否合法购进
待验、合格及不使用物料是否按规定严格管理
药品生产人员是否定期进行健康检查
生产管理
批生产记录是否及时按规定填写
批记录是否能如实和全面反映整个生产过程
生产用设备、容器是否有正确的状态标志
制药企业全套检验记录(DOC)(完整资料).doc

此文档下载后即可编辑********药业有限公司物料检验记录纸编码:XJJL/QC00102版本:A/0检验人:复核人:性状、外观、重(装)量差异检验原始记录编码:XJJL/QC00202版本:A/0检验人:复核人:崩解时限(溶散时限)检验原始记录编码:XJJL/QC00302 版本:A/0检验人:复核人:水分测定检验原始记录编码:XJJL/QC00402 版本:A/0检验人:复核人:卡尔费休水分测定检验原始记录编码:XJJL/QC00502 版本:A/0检验人:复核人:重金属检验原始记录编码:XJJL/QC00602 版本:A/0检验人:复核人:砷盐检查法原始记录编码:XJJL/QC00702 版本:A/0检验人:复核人:生物显微镜检验原始记录编码:XJJL/QC00802版本:A/0检验人:复核人:薄层色谱法检验原始记录编码:XJJL/QC00902版本:A/0检验人:复核人:微生物限度检验原始记录编码:XJJL/QC01002版本:A/0检验人:复核人:高效液相色谱法原始记录编码:XJJL/QC01102版本:A/0检验人:复核人:气相色谱法原始记录编码:XJJL/QC01202 版本:A/0检验人:复核人:红外鉴别原始记录编码:XJJL/QC01302版本:A/0原子吸收分光光度法检验原始记录编码:XJJL/QC01402版本:A/0检验人:复核人:紫外-可见分光光度法检验原始记录编码:XJJL/QC01502版本:A/0复核人:陕西香菊药业集团有限公司物料检验报告单编码:XJJL/QC01602 检字()号陕西香菊药业集团有限公司半成品检验报告单编码:XJJL/QC01702 检字()号陕西香菊药业集团有限公司成品检验报告书编码:XJJL/QC01802 版本:A/0检品编号:报告单号:验:物料检验台账编码:XJJL/QC01902 版本:A/0外包装材料检验台账编码:XJJL/QC02002 版本:A/0编码:XJJL/QC02102 版本:A/0化学试剂分类收发台账编码:XJJL/QC02202 版本:A/0仪器使用情况记录编码:XJJL/QC02302 版本:A/0仪器维护保养记录编码:XJJL/QC02402 版本:A/0计量器具使用情况记录编码:XJJL/QC02502 版本:A/0对照品、标准品、对照药材分类收发台账编码:XJJL/QC02602 版本:A/0滴定液、标准液、检定菌分发记录编码:XJJL/QC02702 版本:A/0。
药品生产企业GMP质量检验记录管理规定

质量检验记录管理规定建立检验记录的书写管理规程,规范记录的书写,保证原始性、真实性、准确性。
第一条:记录的书写要求1记录每页都应该签名和日期,记录应字迹清晰、内容真实、数据完整,记录及时不得超前和回忆记录,内容填写齐全、页码连续,不得留有空格,如无内容填时要用“─”表示,并签名,在每页的右上角标明连续的页码。
2应当在执行操作后立刻在所提供的空白处用不褪色墨水笔填写,不得使用铅笔和涂改液,所有的评论、注释均应使用不褪色墨水笔修改。
3记录应包括样品的基本信息及溶液的制备过程,这些信息要能表明分析人员是按照分析方法规定的程序进行的,所出现的偏差应及时记录。
对于开发的新方法,记录的内容能使其他人清楚地理解,过程的描述要能准确并条理清晰。
4检查数据时发现数据有错误可以划一条横线并写上日期和签名,并写上正确的,检验室对任何一次重新称样都要说明足够的理由。
5附在检验记录中的图谱、表格等,应在图谱和表格上写明检验记录的名称、批号及页码,以防止掉落还能找到相应的位置。
第二条:记录的内容1检测程序(如所用SOP编码)和简要的步骤2标准品的信息,包括:批号、来源、纯度/含量、有效期等。
3检测样品信息,包括:生产厂家、批号。
4检测仪器信息,包括:仪器型号、仪器编号、校验有效期。
5对于仪器的校验和使用记录(比如:PH计、卡尔费休滴定仪与天平),需要记录仪器的编号。
6所有的溶液、稀释液的配制应清楚记录,标准与样品的称重及稀释至最后的浓度。
例如:20mg→100mL;稀释10mL→100mL。
7如果使用已经配制的溶液来制备溶液,应在检验记录里写明溶液的来源索引,以便查找。
8重量与浓度应标上正确的单位,并有计算的公式和举例。
9记录溶液的PH值。
10色谱柱信息(如有),包括型号,编号等。
11环境温湿度(如有需要)。
第三条:检查项下各项书写细则1性状:记录样品的色泽、外观、嗅味,应根据实验中观察到的情况如实描述药品的外观,不可照抄标准上的规定。
药品生产企业日常监督检查跟踪检查记录(片剂、原料药、硬胶囊)

3、该公司按所制订的SOP要求,对空调系统、水系统的日常运行进行记录,并按SOP规定维护、保养;
4、该公司已开展产品年度质量回顾工作,对水系统、环境监测、稳定性试验等进行了趋势分析;公司建立了投诉处理及产品召回流程,明确了产品召回或产品撤回的职责和行动方案。
2019年8月23日市局飞行检查和委托检验专项检查中发现的一项缺陷(易制毒试剂存放现场无易制毒试剂清单),已整改完毕。
现场检查时发现严重缺陷0项、主要缺陷0项,发现一般缺陷0项。
检查中发现的缺陷问题
严重缺陷:无
主要缺陷:无
一般缺陷:无
需要说明的问题:
本次检查现场发现的缺陷不代表公司存在的所有问题,公司应举一反三、持续改进、迅速整改。
附件1
药品生产企业现场检查报告
企业名称
******生物制药股份有限公司
检查范围
**胶囊、**霉素片
地 址
法人
企业负责人
检查部门
**区市场监督管理局
检查
类别
√日常监管 √基本药物 □特殊药品 □中药生产 □GMP跟踪检查
□飞行检查 □委托检验 □委托生产 □不良反应 √市局飞行检查复查、三年内新上市品种
5、根据现场检查该公司生产车间、物料仓库的情况,未见有公司自有品种外的物料、标识物,据公司负责人介绍,该公司无受托或委托生产的情况。
6、该公司近2年未发现违法、违规行为。
严重缺陷:未发现。
主要缺陷:未发现。
一般缺陷:
1、***口服固体制剂车间洁净区器具存放间周转桶“已清洁”标识脱落;(第87条)
制药企业全套检验记录(DOC)

********
药业有限公司
物料检验记录纸
编码:XJJL/QC00102版本:A/0
检品名称
请验单位
批号
批量
检口口编号
检品数量
检验目的
检验项目
收验日期
报告日期
检验依据
性状、外观、重ቤተ መጻሕፍቲ ባይዱ装)量差异检验原始记录
编码:
样品名称
批号
样品编号
规格
数量
检验日期
温度
相对湿度
报告日期
检验依据
□中国药典2010年版部附录()
□其他
性状
外观
重
(装)
量
差
异
天平型号
仪器编号
检验项目
□重量差异
□装量差异□最低装量:
标小量
差异限度
±%
g〜g
±%
g〜g
实测结果
平均重量
不少于标示装量的%
标准规定
超出重量差异限度的不得多于2份,
并不得有1份超出限度1倍。
超出装量差异限度的不得 多于2份,并不得有1份超 出限度1倍。
结论
□(均)符合规定口(均)不符合规定
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
制药企业全套检验记录
背景
制药企业作为医药行业的重要组成部分,需要对其生产的药品进行严格的检验,以确保药品的质量和安全性。
本文档旨在介绍制药企业进行全套检验时需要记录的内容和相关流程。
检验内容
原材料检验
制药企业生产药品所需的原材料需要进行检验,确保其符合相关标准和规定。
原材料检验应包括以下内容:
•检查原材料的外观、颜色、气味等
•检验原材料的纯度和含量
•检验原材料是否存在重金属、农药等有害物质
在制品检验
在制品检验是指在药品生产过程中需要进行的检验,以确保生产过程中的质量
控制得到有效实施。
在制品检验应包括以下内容:
•检查在制品外观是否正常
•检验在制品的含量和纯度是否符合要求
•检验在制品中是否存在杂质等不良物质
成品检验
成品检验是指对制作成功的药品进行检验,以确保药品符合规定的质量标准。
成品检验应包括以下内容:
•检查成品的外观、气味、口感等
•检验成品中的含量和纯度是否符合要求
•检验成品中是否存在杂质等不良物质
检验记录
制药企业进行全套检验时需要进行详细的记录,各类型记录的主要内容如下:
原材料检验记录
原材料检验记录应包括以下内容:
•原材料名称、规格、批号等基本信息
•检验时间和地点
•检验方法和结果
•检验员及其资质证书信息
•药品质量标准
在制品检验记录
在制品检验记录应包括以下内容:
•在制品名称、规格、生产批次等基本信息
•检验时间和地点
•检验方法和结果
•检验员及其资质证书信息
•药品质量标准
成品检验记录
成品检验记录应包括以下内容:
•成品名称、规格、生产批次等基本信息
•检验时间和地点
•检验方法和结果
•检验员及其资质证书信息
•药品质量标准
检验流程
制药企业进行全套检验的流程应包括以下步骤:
1.原材料检验
2.制剂检验
3.包装材料检验
4.成品检验
每一步的检验内容和记录均需严格按照标准和规定执行,并由专业人员进行管理和操作。
总结
制药企业进行全套检验对于保证药品的质量和安全性具有重要作用。
在检验过程中需要注意各项检验内容和相关要求,并进行详细的检验记录,以确保药品质量得到有效保障。