(精选课件)进行性肌营养不良
合集下载
进行性肌营养不良症课件

进行性肌营养不良症课件
目录
• 疾病概述 • 临床表现及病情发展 • 诊断及鉴别诊断 • 治疗及干预措施 • 预防及控制策略 • 研究进展和未来展望
01
疾病概述
Chapter
定义和分类
定义
进行性肌营养不良症(PMD)是一类缓慢进行性 加重的肌肉萎缩、无力,直至最终失去运动功能的 神经肌肉疾病。
分类
主要表现为四肢近端无力、肌肉萎缩等, 但起病年龄较早,病情进展较快,常伴有 肺部感染等症状。
多发性肌炎
周围神经病
是一种自身免疫性疾病,主要表现为四肢 近端无力、肌肉疼痛、皮疹等症状,但肌 肉萎缩不明显,且常伴有皮疹等症状。
主要表现为四肢远端无力、肌肉萎缩等, 但起病年龄较早,病情进展较快,常伴有 感觉异常等症状。
产前诊断
通过基因检测技术,对怀孕期间的胎儿进行诊断,确认是否有进行性肌营养不 良症,以便尽早采取众意识
通过宣传和教育,提高公众对进行性肌营养不良症的认识和关注,促进早期发现 和治疗。
建立支持网络
为患者及其家庭提供心理、社会和医疗支持,建立互助网络,帮助他们更好地应 对疾病。
临床病史
体格检查
实验室检查
电生理检查
肌肉活检
询问患者的肌肉无力 、萎缩等症状出现的 时间、发展速度及伴 随症状等。
检测血清肌酶谱、肌 红蛋白等指标,评估 肌肉损伤程度。
通过取部分肌肉组织 进行病理学检查,确 诊进行性肌营养不良 症。
鉴别诊断
先天性肌营养不良
脊髓性肌萎缩症
与进行性肌营养不良相似,但起病年龄较 早,病情进展较慢,可伴有智力低下、心 脏异常等症状。
Chapter
早期表现
走路延迟
进行性肌营养不良症患者在早期 可能会出现走路延迟,相较于同 龄儿童,可能需要更长时间才能
目录
• 疾病概述 • 临床表现及病情发展 • 诊断及鉴别诊断 • 治疗及干预措施 • 预防及控制策略 • 研究进展和未来展望
01
疾病概述
Chapter
定义和分类
定义
进行性肌营养不良症(PMD)是一类缓慢进行性 加重的肌肉萎缩、无力,直至最终失去运动功能的 神经肌肉疾病。
分类
主要表现为四肢近端无力、肌肉萎缩等, 但起病年龄较早,病情进展较快,常伴有 肺部感染等症状。
多发性肌炎
周围神经病
是一种自身免疫性疾病,主要表现为四肢 近端无力、肌肉疼痛、皮疹等症状,但肌 肉萎缩不明显,且常伴有皮疹等症状。
主要表现为四肢远端无力、肌肉萎缩等, 但起病年龄较早,病情进展较快,常伴有 感觉异常等症状。
产前诊断
通过基因检测技术,对怀孕期间的胎儿进行诊断,确认是否有进行性肌营养不 良症,以便尽早采取众意识
通过宣传和教育,提高公众对进行性肌营养不良症的认识和关注,促进早期发现 和治疗。
建立支持网络
为患者及其家庭提供心理、社会和医疗支持,建立互助网络,帮助他们更好地应 对疾病。
临床病史
体格检查
实验室检查
电生理检查
肌肉活检
询问患者的肌肉无力 、萎缩等症状出现的 时间、发展速度及伴 随症状等。
检测血清肌酶谱、肌 红蛋白等指标,评估 肌肉损伤程度。
通过取部分肌肉组织 进行病理学检查,确 诊进行性肌营养不良 症。
鉴别诊断
先天性肌营养不良
脊髓性肌萎缩症
与进行性肌营养不良相似,但起病年龄较 早,病情进展较慢,可伴有智力低下、心 脏异常等症状。
Chapter
早期表现
走路延迟
进行性肌营养不良症患者在早期 可能会出现走路延迟,相较于同 龄儿童,可能需要更长时间才能
进行性肌营养不良症患者的护理PPT课件
定期随访
定期与医生沟通,进行身体状况的评估和必要的 检查。
早期发现问题,有助于及时处理。
何时寻求帮助?
何时寻求帮助? 症状加重
如患者出现明显的肌肉无力加重、呼吸困难 等,应立即就医。
及时的医疗干预可以避免严重后果。
何时寻求帮助? 心理问题
如果患者供有效的帮助。
心理健康对患者的整体康复至关重要。
如何进行护理?
如何进行护理?
个性化护理计划
根据患者的具体情况制定个性化的护理计划,定 期评估和调整。
护理计划应包括物理治疗、营养指导等。
如何进行护理?
日常护理
协助患者进行日常生活活动,包括起床、洗漱、 穿衣等。
使用辅助设备可以提高患者的自理能力。
如何进行护理?
持续教育对提高护理质量至关重要。
护理团队的角色
患者与家属沟通
与患者及其家属保持良好的沟通,提供必要的支 持和信息。
良好的沟通有助于增强患者的依从性与信任感。
谢谢观看
专业护理可以帮助患者维持日常生活能力, 提高生活质量。
通过适当的护理和康复,可以减缓症状的进 展。
为何需要专业护理? 预防并发症
有效的护理能够预防如肺部感染、关节挛缩 等并发症的发生。
定期监测和早期干预是关键。
为何需要专业护理? 心理支持
患者常常面临心理上的压力与焦虑,专业护 理团队可以提供心理支持。
进行性肌营养不良症患者的护理
演讲人:
目录
1. 什么是进行性肌营养不良症? 2. 为何需要专业护理? 3. 如何进行护理? 4. 何时寻求帮助? 5. 护理团队的角色
什么是进行性肌营养不良症?
什么是进行性肌营养不良症?
定义
进行性肌营养不良症健康宣教PPT课件

为什么要关注进行性肌营养不 良症?
为什么要关注进行性肌营养不良症? 影响生活质量
肌营养不良症会导致运动能力下降,影响日 常生活和独立性。
早期干预可以改善患者的生活质量。
为什么要关注进行性肌营养不良症? 心理健康
患者常常面临焦虑、抑郁等心理健康问题。
理解疾病及其影响有助于心理支持和治疗。
为什么要关注进行性肌营养不良症? 家庭影响
这种疾病不仅影响患者,也影响家庭成员的 生活和情感状态。
家庭支持在患者康复中起重要作用。
何时及如何进行诊断?
何时及如何进行诊断? 早期症状
常见早期症状包括行走困难、肌肉无力等。
家长应关注孩子的运动能力变化。
何时及如何进行诊断? 诊断方法
通过临床评估、基因检测和肌肉活检Байду номын сангаас方法进行 确诊。
及早诊断有助于制定合理的治疗方案。
进行性肌营养不良症健康宣教
演讲人:
目录
1. 什么是进行性肌营养不良症? 2. 为什么要关注进行性肌营养不良症? 3. 何时及如何进行诊断? 4. 如何进行管理与治疗? 5. 向谁寻求帮助?
什么是进行性肌营养不良症?
什么是进行性肌营养不良症? 定义
进行性肌营养不良症是一组遗传性疾病,导致肌 肉逐渐弱化和萎缩。
何时及如何进行诊断? 专业咨询
建议患者及家属咨询专业医生,获取准确的信息 和指导。
医疗团队包括医生、物理治疗师、心理医生等。
如何进行管理与治疗?
如何进行管理与治疗? 物理治疗
物理治疗可以帮助维持肌肉功能,减缓疾病 进展。
定期的运动和康复训练是关键。
如何进行管理与治疗? 药物治疗
某些药物可以帮助改善症状,延缓肌肉损伤 。
《进行性肌营养不良》课件
瑜伽
瑜伽可以协助患者放松肌肉、缓解压力、增强灵活 性。
总结说明
患者需要休息
进行性肌营养不良的患者需要充足的休息和睡眠,帮助他们更好地处理症状和并发症。
医生建议
患者和家庭成员应该与专业医生保持沟通,了解最佳的治疗和护理方式。
协作治疗
进行性肌营养不良往往需要团队合作来全面治疗患者。
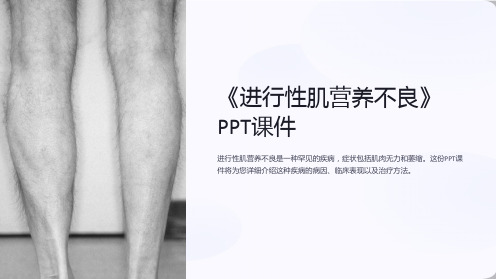
肌肉萎缩
受累肌肉可能会逐渐萎缩,因为它们没有正常的运 动和使用。
行动不便
进行性肌养不良会导致肌肉无力和萎缩,使病人 行动不便。
D iag nosis
1 肌肉活检
医生可能会取一个小肌肉组织样本,用显微镜检查它们是否表现出进行性肌营养不良的 迹象。
2 电磁信号测试
医生可以将电极放在表现出症状的肌肉上,从而检查肌肉活动和电信号是否异常。
病因分析
1
基因突变
进行性肌营养不良是由基因突变引起的,这些基因编码肌肉细胞中的蛋白质。
2
毒素作用
有些外部毒素也会引起进行性肌营养不良,如酒精和某些农药。
3
其他因素
此外,还有其他因素可以影响这种疾病在个体中的表现,如基因表达、环境、代 谢和免疫功能。
临床表现
肌肉无力
患者的肌肉无力可能会逐渐加重,直到影响生活质 量。
治疗方法
1
药物治疗
一些药物,如皮质类固醇和免疫抑制剂,
物理疗法
2
可以帮助减轻肌肉无力和萎缩。
物理疗法,如电动力治疗和康复运动,
可以帮助提高肌肉的力量和灵活性。
3
睡眠和饮食管理
维持充足的睡眠和健康的饮食也可以有 助于缓解症状和提高生活质量。
预后与护理
康复医疗
康复医疗是处理进行性肌营养不良的基本方式。
进行性肌营养不良症健康教育PPT

谢谢观看
家族史是一个重要的风险因素,许多病例是遗传 性。
谁会受到影响?
谁会受到影响? 受影响的人群
主要影响男性,尤其是儿童和青少年,女性 相对少见。
不同类型的肌营养不良症在性别上的影响也 有所不同。
谁会受到影响? 遗传模式
大多数类型以X连锁隐性遗传方式遗传,父母 可能是携带者。
了解家族历史有助于识别潜在风险。
进行性肌营养不良症健康教 育
演讲人:
目录
1. 什么是进行性肌营养不良症? 2. 谁会受到影响? 3. 何时需要就医? 4. 如何管理和治疗? 5.
什么是进行性肌营养不良症? 定义
进行性肌营养不良症是一类遗传性肌肉疾病,特 征为肌肉逐渐无力和萎缩。
保持适当的锻炼和营养,增强体能和肌肉力 量。
适合的锻炼计划应由专业人士制定。
如何管理和治疗? 心理支持
心理支持和家庭支持对患者的心理健康至关 重要。
可以考虑心理咨询或加入支持小组。
为什么要重视进行性肌营养 不良症?
为什么要重视进行性肌营养不良症? 提高认知
提高对该疾病的认知,有助于早期发现和干预。
谁会受到影响? 发病年龄
通常在儿童期或青少年期发病,早期发病往 往预后较差。
延迟发病可能导致较好的生活质量。
何时需要就医?
何时需要就医?
症状表现
出现肌肉无力、行走困难、跌倒频繁等症状时, 应及时就医。
早期诊断有助于采取干预措施,改善生活质量。
何时需要就医?
体检与评估
医生会进行体格检查、血液检测及基因检测等, 以确认诊断。
此病主要影响青少年和儿童,影响运动能力和生 活质量。
什么是进行性肌营养不良症? 类型
主要类型包括杜氏肌营养不良、贝克肌营养不良 等,各种类型的症状和进展速度有所不同。
进行性肌营养不良症护理业务学习PPT课件

进行性肌营养不良症护理业务学 习
演讲人:
目录
1. 什么是进行性肌营养不良症? 2. 为什么进行性肌营养不良症需要护理? 3. 如何进行护理? 4. 护理中的挑战与对策 5. 未来的护理展望
什么是进行性肌营养不良症?
什么是进行性肌营养不良症?
定义
进行性肌营养不良症是一组遗传性疾病,主要影 响肌肉的力量和功能。
护理的主要目标是促进患者的独立性,减少 并发症,并提供心理支持。
这包括物理治疗、营养支持和心?
社会支持
患者及其家庭需要社会支持,包括医疗资源 、教育和社区服务。
有效的社会支持可以提高患者的生活质量, 减轻家庭负担。
如何进行护理?
如何进行护理?
评估
定期评估患者的肌肉功能、日常生活能力和心理 状态,以制定个性化护理计划。
有效的团队合作可以提高护理效率,改善患 者的综合管理。
未来的护理展望
未来的护理展望
研究方向
未来的研究将集中在新的治疗方法、早期筛查和 个性化护理方案的开发上。
包括基因治疗和干细胞研究等前沿技术。
未来的护理展望
技术应用
先进技术如远程医疗和智能护理设备将有助于提 高护理的可及性和效果。
这些技术可以提供实时监测和支持,改善患者的 生活质量。
未来的护理展望
政策支持
呼吁社会对进行性肌营养不良症患者的关注,推 动相关政策的制定和实施。
包括医疗保险、教育支持和社区服务等方面的政 策。
谢谢观看
这类疾病通常表现为肌肉逐渐萎缩和无力,影响 日常活动。
什么是进行性肌营养不良症?
病因
大多数类型的进行性肌营养不良症是由基因突变 引起的,这些突变影响肌肉纤维的结构和功能。
常见的类型包括杜氏肌营养不良症和贝克肌营养 不良症。
演讲人:
目录
1. 什么是进行性肌营养不良症? 2. 为什么进行性肌营养不良症需要护理? 3. 如何进行护理? 4. 护理中的挑战与对策 5. 未来的护理展望
什么是进行性肌营养不良症?
什么是进行性肌营养不良症?
定义
进行性肌营养不良症是一组遗传性疾病,主要影 响肌肉的力量和功能。
护理的主要目标是促进患者的独立性,减少 并发症,并提供心理支持。
这包括物理治疗、营养支持和心?
社会支持
患者及其家庭需要社会支持,包括医疗资源 、教育和社区服务。
有效的社会支持可以提高患者的生活质量, 减轻家庭负担。
如何进行护理?
如何进行护理?
评估
定期评估患者的肌肉功能、日常生活能力和心理 状态,以制定个性化护理计划。
有效的团队合作可以提高护理效率,改善患 者的综合管理。
未来的护理展望
未来的护理展望
研究方向
未来的研究将集中在新的治疗方法、早期筛查和 个性化护理方案的开发上。
包括基因治疗和干细胞研究等前沿技术。
未来的护理展望
技术应用
先进技术如远程医疗和智能护理设备将有助于提 高护理的可及性和效果。
这些技术可以提供实时监测和支持,改善患者的 生活质量。
未来的护理展望
政策支持
呼吁社会对进行性肌营养不良症患者的关注,推 动相关政策的制定和实施。
包括医疗保险、教育支持和社区服务等方面的政 策。
谢谢观看
这类疾病通常表现为肌肉逐渐萎缩和无力,影响 日常活动。
什么是进行性肌营养不良症?
病因
大多数类型的进行性肌营养不良症是由基因突变 引起的,这些突变影响肌肉纤维的结构和功能。
常见的类型包括杜氏肌营养不良症和贝克肌营养 不良症。
进行性肌营养不良症科普讲座课件

可能出现的并发症包括呼吸问题和心脏病。
定期监测和早期干预可以降低并发症风险。
如何诊断进行性肌营养不良症 ? Nhomakorabea如何诊断进行性肌营养不良症?
临床评估
医生会通过病史询问和体格检查来初步评估 患者的症状。
肌肉力量测试和功能评估是重要的诊断步骤 。
如何诊断进行性肌营养不良症?
辅助检查
血液检查、肌肉活检和基因检测可以帮助确 认诊断。
这些疾病影响肌肉的结构和功能,导致运动能力 下降。
什么是进行性肌营养不良症?
分类
常见的类型包括杜氏肌营养不良、贝克肌营养不 良等。
不同类型的疾病可能在症状和发病年龄上有所不 同。
什么是进行性肌营养不良症? 发病机制
这些疾病通常是由于特定基因的突变导致肌肉细 胞无法正常生成所需的蛋白质。
这一机制导致肌肉细胞逐渐丧失功能。
谢谢观看
进行性肌营养不良症科普讲座
演讲人:
目录
1. 什么是进行性肌营养不良症? 2. 谁会受到影响? 3. 症状和体征是什么? 4. 如何诊断进行性肌营养不良症? 5. 如何管理和治疗进行性肌营养不良症?
什么是进行性肌营养不良症?
什么是进行性肌营养不良症?
定义
进行性肌营养不良症是一组遗传性疾病,主要特 征是肌肉逐渐无力和萎缩。
遗传咨询可以帮助评估风险。
症状和体征是什么?
症状和体征是什么? 初期症状
初期症状可能包括肌肉无力、运动困难和频繁跌 倒。
这些症状通常在日常活动中逐渐显现。
症状和体征是什么? 发展过程
随着病情进展,患者可能会出现肌肉萎缩、关节 挛缩和心肺功能受损。
这会显著影响生活质量和日常活动能力。
症状和体征是什么? 并发症
定期监测和早期干预可以降低并发症风险。
如何诊断进行性肌营养不良症 ? Nhomakorabea如何诊断进行性肌营养不良症?
临床评估
医生会通过病史询问和体格检查来初步评估 患者的症状。
肌肉力量测试和功能评估是重要的诊断步骤 。
如何诊断进行性肌营养不良症?
辅助检查
血液检查、肌肉活检和基因检测可以帮助确 认诊断。
这些疾病影响肌肉的结构和功能,导致运动能力 下降。
什么是进行性肌营养不良症?
分类
常见的类型包括杜氏肌营养不良、贝克肌营养不 良等。
不同类型的疾病可能在症状和发病年龄上有所不 同。
什么是进行性肌营养不良症? 发病机制
这些疾病通常是由于特定基因的突变导致肌肉细 胞无法正常生成所需的蛋白质。
这一机制导致肌肉细胞逐渐丧失功能。
谢谢观看
进行性肌营养不良症科普讲座
演讲人:
目录
1. 什么是进行性肌营养不良症? 2. 谁会受到影响? 3. 症状和体征是什么? 4. 如何诊断进行性肌营养不良症? 5. 如何管理和治疗进行性肌营养不良症?
什么是进行性肌营养不良症?
什么是进行性肌营养不良症?
定义
进行性肌营养不良症是一组遗传性疾病,主要特 征是肌肉逐渐无力和萎缩。
遗传咨询可以帮助评估风险。
症状和体征是什么?
症状和体征是什么? 初期症状
初期症状可能包括肌肉无力、运动困难和频繁跌 倒。
这些症状通常在日常活动中逐渐显现。
症状和体征是什么? 发展过程
随着病情进展,患者可能会出现肌肉萎缩、关节 挛缩和心肺功能受损。
这会显著影响生活质量和日常活动能力。
症状和体征是什么? 并发症
神经病学-进行性肌营养不良精品PPT教学课件

2020/12/6
9
女性为基Байду номын сангаас携带者,所生男孩约50%发 病,女孩患病者罕见;
有些携带者可有肢体无力、腓肠肌肥大、 血清CK增高等临床表现。患儿多呈明确 家族性,另有1/3患儿由新的基因突变 所致病。
2020/12/6
10
临床表现是:
①患儿均为男性,多在3~5岁发病; 起病隐袭,开始症状多为行走慢,不能
2020/12/6
6
病理
PMD的肌肉基本病理改变为肌纤维坏死 和再生,肌膜核内移,出现肌细胞萎缩 与代偿性增大相嵌分布的典型表现,并 随病情进展这种肌细胞大小差异不断增 加。
2020/12/6
7
肥大肌细胞横纹消失,光镜下呈玻璃样 变;
坏死肌细胞出现空泡增多、絮样变性、 颗粒变性和吞噬现象。
约1/3患儿智力发育迟缓。
2020/12/6
19
一般无消化道症状,少见的并发症为急 性胃扩张;
患者多在25~30 岁以前死于呼吸道感染、 心力衰竭或消耗性疾病;
2020/12/6
20
③本型的病情是PMD中最严重的,其严重程度 与患儿家族中遗传代数成反比,即家族中受累 代数越多,病情越轻,最重的是散发病例,预 后不良;
正常跑步,容易跌倒; 肌无力自躯干和四肢近端开始缓慢进展,
下肢重与上肢;
2020/12/6
11
鸭步:
骨盆带肌肉无力,肌张力减低,由于髂 腰肌和股四头肌无力,而登楼及蹲位站 立困难,进而腰椎前凸;因骨盆带肌无 力而行走时向两侧摇摆。
2020/12/6
12
Gower征(攀登起立征):
由于腹肌和髂腰肌的无力,仰卧站立时, 患儿必须先转为俯卧位,然后以双手支 撑双足背、膝部等处顺次攀附,方能起 立。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
大多数伴心肌损害: 心率不齐,心脏扩大
约30%患儿伴不同程度智障 预后:差!12岁左右不能行走(此点有助于鉴别DMD和BMD), 多数患者20-30岁因呼吸道感染、心力衰竭而死亡
13
DMD型的诊断
14
鉴 别 诊 断1
15
鉴 别 诊 断2
16
鉴 别 诊 断3
格林巴利综合征: 1.概述:是常见的脊神经和周围神经的脱髓鞘疾病。 2.临床表现: 感染性疾病后1-3周,突然出现剧烈的神经根疼痛 进行性上升性对称性麻痹,四肢软瘫,运动障碍,不
堆
病 理:
组织学肌肉病理: 进行性肌纤维坏死 再生 脂肪及结缔组织增生 肌肉无异常代谢产物
3
分类
4
5
辅助检查
1.诊断肌肉疾病中的应用(血清酶学检查):
肌酸激酶(CK)
假肥大型
乳酸脱氢酶(LDH)
异常 见于 远端型
肌酸激酶同工酶(CK-MB)
肢带型
丙氨酸氨基转移酶(ALT)
门冬氨酸氨基转移酶(AST) 在进展期均可轻度升高
肌细胞坏死和功能缺失
病理
9
临床表现
肌肉假性肥大,触之坚韧,以腓肠肌最明显(常为首 发症状之一。
为萎缩肌纤维坏死后被脂肪和结缔组织替代所致
10
Gower征:(为DMD的特征性表现) 腹肌和髂腰肌无力 患儿自仰卧位站立时必须特征性的用手支撑方能站起
11
鸭步:
臀中肌无力致行走时骨盆向两侧 上下摆动
进行性肌营养不良
1
概述
是一组遗传性肌肉变性病 表现为缓慢进行性加重的对称性肌无力、肌
肉萎缩 无感觉障碍 多发生于儿童和青少年 电生理:肌源性损害,神经传导速度正常 组织学:进行性的肌纤维坏死、再生和脂肪
及结缔组织增生
2
病 因:
遗传方式: 染色体显性遗传 染色体隐性遗传 X连锁隐性遗传
走路以脚尖着地,走路慢:
3-5岁隐匿出现骨盆带肌肉无力 下肢伸肌群力量不足
腰椎前凸:
背部伸肌无力所致
马蹄足内翻 翼状肩胛:举臂时肩胛骨内侧远 离胸壁,两肩胛骨呈翼状竖起于背部
12
肌肉无力及萎缩: 从下肢开始,逐渐出现躯干、髋、上肢、肩部肌肉 萎缩,患儿无法站立,甚至卧床不起 后期出现跟腱挛缩,双足下垂
血清肌红蛋白(MB)
2.肌电图检查:
肌源性损害
3.尿检 :
尿酸排出增多,肌酐减低(贝氏型)
4.CT:病变肌肉呈密度减低影
6
辅助检查
5.MRI:可见受累肌肉不同程度的“蚕食现象” 受累肌群被脂肪代替改变 能为临床选取准确活检部位提供定位
6.肌肉活检: 肌肉的坏死、再生、间质脂肪和结缔组织增生 抗肌萎缩蛋白缺失
同程度的感觉障碍;腱反射减弱或消失,颅神经症状。 3.检查: 脑脊液检查:出现典型的蛋白质增加而细胞数正常。 4.预后:多数可恢复。
17
治疗
18
7.DNA检测: Xp21基因突变
8.心电图:排除心肌的损伤 9.智力检测:DMD和BMD患者应做
7
Duchenne型肌营养不良
1.概述:是临床最常见类型,儿童多发
为X性连锁隐性遗传(只有男婴患病,女性为致病基因携带者) 发病年龄:3-5岁
2.病理: 基因缺失
肌细胞内抗肌萎缩蛋白缺失
肌细胞膜不稳定
约30%患儿伴不同程度智障 预后:差!12岁左右不能行走(此点有助于鉴别DMD和BMD), 多数患者20-30岁因呼吸道感染、心力衰竭而死亡
13
DMD型的诊断
14
鉴 别 诊 断1
15
鉴 别 诊 断2
16
鉴 别 诊 断3
格林巴利综合征: 1.概述:是常见的脊神经和周围神经的脱髓鞘疾病。 2.临床表现: 感染性疾病后1-3周,突然出现剧烈的神经根疼痛 进行性上升性对称性麻痹,四肢软瘫,运动障碍,不
堆
病 理:
组织学肌肉病理: 进行性肌纤维坏死 再生 脂肪及结缔组织增生 肌肉无异常代谢产物
3
分类
4
5
辅助检查
1.诊断肌肉疾病中的应用(血清酶学检查):
肌酸激酶(CK)
假肥大型
乳酸脱氢酶(LDH)
异常 见于 远端型
肌酸激酶同工酶(CK-MB)
肢带型
丙氨酸氨基转移酶(ALT)
门冬氨酸氨基转移酶(AST) 在进展期均可轻度升高
肌细胞坏死和功能缺失
病理
9
临床表现
肌肉假性肥大,触之坚韧,以腓肠肌最明显(常为首 发症状之一。
为萎缩肌纤维坏死后被脂肪和结缔组织替代所致
10
Gower征:(为DMD的特征性表现) 腹肌和髂腰肌无力 患儿自仰卧位站立时必须特征性的用手支撑方能站起
11
鸭步:
臀中肌无力致行走时骨盆向两侧 上下摆动
进行性肌营养不良
1
概述
是一组遗传性肌肉变性病 表现为缓慢进行性加重的对称性肌无力、肌
肉萎缩 无感觉障碍 多发生于儿童和青少年 电生理:肌源性损害,神经传导速度正常 组织学:进行性的肌纤维坏死、再生和脂肪
及结缔组织增生
2
病 因:
遗传方式: 染色体显性遗传 染色体隐性遗传 X连锁隐性遗传
走路以脚尖着地,走路慢:
3-5岁隐匿出现骨盆带肌肉无力 下肢伸肌群力量不足
腰椎前凸:
背部伸肌无力所致
马蹄足内翻 翼状肩胛:举臂时肩胛骨内侧远 离胸壁,两肩胛骨呈翼状竖起于背部
12
肌肉无力及萎缩: 从下肢开始,逐渐出现躯干、髋、上肢、肩部肌肉 萎缩,患儿无法站立,甚至卧床不起 后期出现跟腱挛缩,双足下垂
血清肌红蛋白(MB)
2.肌电图检查:
肌源性损害
3.尿检 :
尿酸排出增多,肌酐减低(贝氏型)
4.CT:病变肌肉呈密度减低影
6
辅助检查
5.MRI:可见受累肌肉不同程度的“蚕食现象” 受累肌群被脂肪代替改变 能为临床选取准确活检部位提供定位
6.肌肉活检: 肌肉的坏死、再生、间质脂肪和结缔组织增生 抗肌萎缩蛋白缺失
同程度的感觉障碍;腱反射减弱或消失,颅神经症状。 3.检查: 脑脊液检查:出现典型的蛋白质增加而细胞数正常。 4.预后:多数可恢复。
17
治疗
18
7.DNA检测: Xp21基因突变
8.心电图:排除心肌的损伤 9.智力检测:DMD和BMD患者应做
7
Duchenne型肌营养不良
1.概述:是临床最常见类型,儿童多发
为X性连锁隐性遗传(只有男婴患病,女性为致病基因携带者) 发病年龄:3-5岁
2.病理: 基因缺失
肌细胞内抗肌萎缩蛋白缺失
肌细胞膜不稳定