云南省建设工程规划许可证申请表(空表)
建设用地规划许可证申请表

建设用地规划许可证申请表项目编号:建设单位名称法人代表电话联系人电话建建设项目选址建设项目名称延规选址()号意见书设建设项目位置单规划设计单位规划设计资质位东至:南至:建设项目用地边界西至:北至:m 建筑面积 ? 建筑高度规划总用地面积 ? 规划用地性质建筑基底面积 ? 绿地面积 ?% 建筑总面积 ? 绿地率% 建筑密度容积率机动车停车位地上:个地下:个备注1、《建设用地规划许可证》申请表一份。
2、建设单位书面提出申请办理《建设用地规划许可证》的综合报告(说明申请理由、项目基本情况、用地基本情况、规划设计方案是否符合规划设计条件、其他需说明事项的申报材料统一说明)。
3、申请人应提交法人资格证明(工商营业执照或组织机构法人代码证)(复印件),关于办理《建设用地规划许可证》的法人授权委托书一份、法人和受托人的身份证复印件一份。
应4、在自有土地上进行新建、扩建、改建的建设项目需提供《国有土地使用证》、《房屋所有权证》(复印件)一份。
提 5、建设用地规划条件附图三份。
供 6、经有关部门批准的本年度投资计划,项目批准文件。
的 7、已取得的《建设项目选址意见书》(复印件)及附件、附图(原件)各一份。
文 8、以出让方式取得国有土地使用权的建设项目还需提供建设项目的批准、核准、备案文件和《国有土地使用权出让合同》(复印件)一份及《国有土地拟批准书》。
件9、报州、市城镇规划委员会审议的建设项目需提交州、市城镇规划委员会有明确、同意意见的会议纪要一份。
图 10、扩建或加层的建设项目应提供相关部门的技术鉴定纸 11、除规划确定的城市基础设施等项目外,建设项目需占用其他单位土地或征用集体土地的应提供双方用地协议书和土地管理部门意见。
及12、重要建设工程或大、中型项目应提供可行性报告或项目建议书。
资 13、工业项目、对环境有影响的建设项目需提交相关主管部门的意见。
料14、特殊行业的建设项目应提交行业行政主管部门的批准文件。
建设工程规划许可证(零星)申请表(建制镇个人建房)

平房
西靠
灶间
灶间
南靠
副房
副房
பைடு நூலகம்北靠
户主
人口数
家
庭
成
员
关系
姓名
性别
户口类型
出生年月
申请建房理由:
送审文件、图纸一览表
序号
名称
应送份数
实送份数
要件说明
注意事项(填表前,请仔细阅读下列内容并遵照执行):
一、本表适用于下列建设工程申请《建筑工程规划许可证(零星)》(建制镇个人建房):
建制镇的个人在原住房用地或者经规划调整的个人住房用地范围内,建造居住房屋。
5、属危险房屋的、应提供危房鉴定证明(一份);
6、因建设项目的特殊性需要提交的其它相关材料。
建设工程规划许可证(零星)申请表(建制镇个人建房)
*为必填字段
申请人姓名*
邮政编码*
申请人签名、盖章
地址*
联系电话*
手机号码
原住房位置*
用地性质:国有集体
用地性质:国有集体
修建住房位置*
原有住房
修建住房
类型
占地面积
(平方米)
建筑面积
(平方米)
类型
占地面积
(平方米)
建筑面积
(平方米)
四周界址
楼房
楼房
东靠
二、随申请表应按下列要求送审相关文件、图纸:
1、1/500地形图和建筑平面图,用红色实线标明用地范围(一份);
2、施工图,建造的建筑为3层以上(含3层)的,施工图应由有资质的建筑设计单位设计或者复核(二套);
3、原建制镇宅基地的使用证明(原件和复印件各一份);
4、常住户籍证明(原件和复印件各一份);
建设工程规划许可证申请表(范本)

总建筑面积 (M2):
190000
总建筑基底面积(M2): 8000
建筑名称:
1号楼 2号楼 3号楼
4号楼
5号楼
6号楼
使用功能:
住宅
住宅
住宅
住宅
住宅
住宅
层 数:
11
11
11
11
11
11
建筑基底面积 (M2):
600
600
600
600
600
600
使
地上建筑面积 (M2):
5500 5500 5500
5500 5500
216
停 机动车 车位,其中地面 车位,地下 车位。
车 非机动车 车位,其中地面 车位,地下 车位。 位 地下车库出入口方向:
其 他 要 求
有 关 部 门 意 见
项目总编号:
总关联编号:
续前一页设计指标(本页必须盖章有效)
建筑 名称
使用功能
阳台面 积(M2)
层数
建筑基 地上建 底面积 筑面积 (M2) (M2)
建筑长度(M): 标 建筑宽度(M):
建筑高度(M):
架空层面积 (M2):
阁楼层面积 (M2):
阳台面积(M2):
总地上建筑面积 总地下室面积 总配套设施面 总架空层面积 总阁楼层面积 总阳台面积
(M2):
(M2):
积(M2):
(M2):
(M2):
(M2):
停 机动车 车位,其中地面 车位,地下 车位。 车 非机动车 车位,其中地面 车位,地下 车位。 位 地下车库出入口方向:
5500
用
地下室面积 (M2):
600 600 600
《建设工程规划许可证》(建筑类)申请表

地面 地下 地面 地下
住宅 套型 面积
大于 90 m2 小于 90 m2
总计
户数
建筑面积〔m2
建设单位〔公章 年 月日
填写须知: 根据有关法律规定,
申请人应如实提交有关材料和 反映真实情况,并对申请材料 实质内容的真实性负责。以虚 报、瞒报、造假等不正当手段 取得批准文件的,将依法予以 撤销,并追究相应责任。
章;
2、法人委托书〔加盖建设单位公章及法人章须有法人签名或盖章,法人身份证及经办人身份证〔复
印件;
3、申请报告;
4、《土地证》及其附图、《建设用地规划许可证》及其附图〔以上均为复印件一份;
5、区、市发改委计划立项批文一份〔复印件、房改办批文一份〔复印件;
应 6、原审批的总平面图或设计兰线图一份〔复印件;
6设计单位施工单位及施工图测量单位须认真核实表中各项内容并对由本单位提供的申请材料的内容真实性负责如出现虚报瞒报漏报及造假等情况将通报其上级主管部门按相关规定进行处理
以下内容由 XX 市规划管理局填写 初 审
审 核
审 定
核 发
项目编号:
.
XX 市规划管理局
建设工程规划许可证〔建筑
类申请表
申请单位:
建设单位〔公章
项目名称:
法人代表
联系人
设计单位〔公章
项目负责人 资质证书编号
施工图测绘单位 〔公章
项目负责人 资质证书编号
项目名称
建设性质
建筑结构
建设地址
总投资额 〔万元
项目类型〔请在 选项前方框内
打"√"
□房地产项目
□廉租房项目
迁回建房项目 □其他
□政府投资项目 □非政府投资项目
《建设工程规划许可证》申请表

根据《中华人民共和国城市规划法》、《江苏省实施〈中华人民共和国城市规划法〉办法》之规定,城市规划管理实行由城市规划行政主管部门核发《建设项目选址意见书》、《建设用地规划许可证》、《建设工程规划许可证》制度。
建设单位申请办理《建设工程规划许可证》应报送如下资料:
1、建设单位如实填写《建设工程规划许可证》申请表1份,并在申请表封面建设单位栏加盖单位公章,出示组织机构代码证原件并提供复印件1份。
2、建设项目有效批准文件。
3、1:500或1:1000统一城市坐标系的现势地形图2份。
4、规划设计条件及附图各1份。
5、规划方案审查意见书及批准的方案各1份。
6、出示土地使用权属证明原件,提供复印件1份。
7、建设工程全套施工图2份(电子EORD文档1份)。
8、消防部门书面审查意见书。
9、与申请事项有关的资料。
注:凡提供的复印件必须加盖建设单位公章,并注明“与原件核对无
误”字样。
《建设工程规划许可证》申请表
建设项目
建设单位
主管部门
经办人员
联系电话
联系地址
收件编号年号
许可证号发证日期。
填写样本建设用地规划许可证申请表
〔公章〕
审查人:XX年 月
序号
材料 名 称
类型
备 注
1
申请人身份证证〕;
复印
开发类工程需提交开发 资质.
2
1: 500或1:1000与现状相符的地形图〔有资质的测绘部门〕;
原件
3
国有土地使用权出让合同〔出让方式〕;建设工程选址意见
书〔划拨方式〕
复印
4
建设工程批准、核准、备案文件;
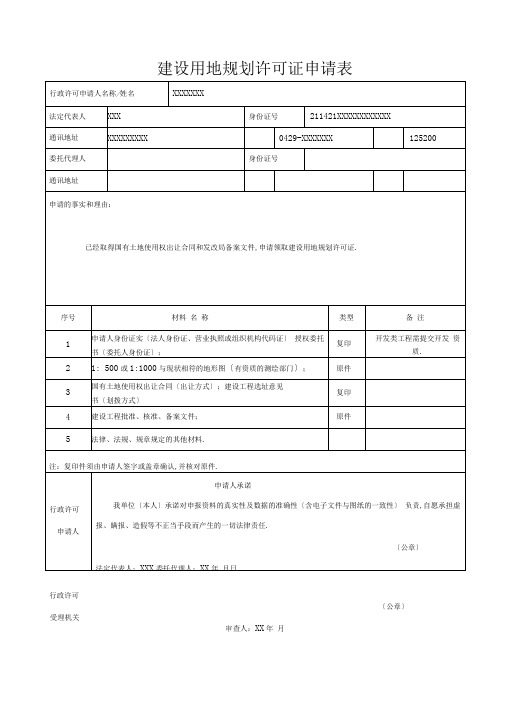
建设用地规划许可证申请表
行政许可申请人名称/姓名
XXXXXXX
法定代表人
XXX
身份证号
211421XXXXXXXXXXXX
通讯地址
XXXXXXXXX
0429-XXXXXXX
125200
委托代理人
身份证号
通讯地址
申请的事实和理由:
已经取得国有土地使用权出让合同和发改局备案文件,申请领取建设用地规划许可证.
原件
5
法律、法规、规章规定的其他材料.
注:复印件须由申请人签字或盖章确认,并核对原件.
行政许可
申请人
申请人承诺
我单位〔本人〕承诺对申报资料的真实性及数据的准确性〔含电子文件与图纸的一致性〕 负责,自愿承担虚报、瞒报、造假等不正当手段而产生的一切法律责任.
〔公章〕
法定代表人:XXX委托代理人:XX年 月日
建设用地规划许可证申请表

□国有土地使用权划拨文件(纸质,复印件1份,加盖建设单位公章,验原件)
□项目建设用地位置界限图(纸质,原件2份,加盖规划技术服务中心资质章)
□法律、法规等定的其他材料:
建
设
单
位
承
诺
本单位对所提交的材料的真实性及其内容的准确性、合法性、规范性负责,如有虚假和错误,本单位将承担由此引起的一切后果。
用地单位:(盖章)
建设用地规划许可证申请表
用地单位
单位地址
法人代表
联系电话
用地项目名称
用地项目位置
用地面积
建设规模
联系人
电话
申请类别
□新建□补办□变更□挂失□临时用地□其他:
报送材料目录:
□《建设用地规划许可证》申请表1份
□建设项目批准、核准、备案文件(纸质,复印件1份,加盖建设单位公章,验原件)
□建设项目选址意见书(纸质,复印件1份,加盖建设单位公章,验原件)
《建设工程规划许可证》申请表

年月日
建设单位:
我单位保证所提供的文件资料真实
有效并承担相关法律责任
单位地址项目名称:申请建设规模:
申请项目地址:(区)旗
设计单位(施工图):
我单位保证所提供的文件资料真实
有效并承担相关法律责任
联系人:电话:单位公章
申请性质:新报()
补办()
延期()
变更()
注意事项:
1、本表由建设单位、设计单位填写并加盖公章(建设单位一栏要填写单位全称或法定简称)。
2、请在申请性质一栏的()中划√表明申报内容。
3、按申请资料要求备齐文件、图纸装订成册后方可城市规划行政主管部门申请,否则不予受理。
4、城市规划行政主管部门受理申请后发出受理通知书,请妥善保存,并凭受理通知书原件的规定的期限内领取审批文件。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
规划行政主管部门内部业务会签意见
城市
规划行政主管部门建设工程规划审批意见
签发人: (单位盖章)年月日
附图附件名称
(备注:证收编号数字共15位。其中:前6位数号码按照《中华人民共和国行政区划代码》Байду номын сангаасGB/T2260)执行;7—10位数号码代表证书发放年份;11—15位数号码代表证书发放序号。)
编号:建字第号
云南省
建设工程规划许可证
申请表
申请单位:
项目名称:
申报日期:
云 南 省 建 设 厅 监 制
云南省城市勘察测量规划协会印制
项目总编号
案卷编号
委托人
联系电话
法人代表
联系电话
建设单位名称
项目名称
建设地点
建设用地性质
建设用地规划许可证号
设计单位(资质)
项目简介
申报材料
备注
郑重承诺
我单位已阅知行政许可告知书等说明,并承诺对申报材料的真实性、数据的准确性(含相关电子文件与图纸的一致性)负责,自愿承担虚报、瞒报、造假等不正当手段而产生的一切法律责任。
(申请单位盖章)
二〇一年月日
工程名称
栋数
结构
层数
建筑面积(m2)
基底面积
总面积
总占地面积(m2)
工程总投资(万元)
总建筑面积(m2)
地下(m2)
地上(m2)
城市规划行政主管部门初审意见
经办(处、科、室)负责人:
年月日