药品试液配制含量均匀度检查法
阿莫西林克拉维酸钾干混悬剂(14∶1)的含量均匀度测定和有关物质检查

阿莫西林克拉维酸钾干混悬剂(14∶1)的含量均匀度测定和有关物质检查作者:李静玲李瑞明冯鹏胡建楣林小凤范佩冰来源:《中国医药导报》2013年第07期[摘要] 目的对阿莫西林克拉维酸钾干混悬剂(14∶1)的含量均匀度和有关物质进行了研究,用来评价和确定制剂使用中的安全性及药物本身的内在质量。
方法通过对制备得到的阿莫西林克拉维酸钾干混悬剂含量均匀度的测定和有关物质考察,从而确定阿莫西林克拉维酸钾干混悬剂质量的可控性。
结果通过本文中所用方法对阿莫西林克拉维酸钾干混悬剂(14∶1)中阿莫西林、克拉维酸钾的含量均匀度和有关物质测定结果说明制剂质量合格,所以在制剂使用中阿莫西林克拉维酸钾干混悬剂能确保其安全性。
结论阿莫西林克拉维酸钾干混悬剂(14∶1)中阿莫西林、克拉维酸钾的含量均匀度和有关物质的测定均能符合制剂要求。
[关键词] 阿莫西林;克拉维酸钾;含量均匀度;有关物质[中图分类号] R927.11 [文献标识码] A [文章编号] 1673-7210(2013)03(a)-0123-03阿莫西林(Amoxicillin)(C16H19N3O5S)是一种最常用的青霉素类广谱β-内酰胺类抗生素,半衰期约为61.3 min。
阿莫西林杀菌作用强,穿透细胞膜的能力也强,是目前应用较为广泛的口服青霉素之一,其制剂有胶囊、片剂、颗粒剂、分散片等。
单独用阿莫西林时容易产生耐药性而降低药物的疗效;克拉维酸钾(C8H9NO5)仅有微弱的抗菌活性,但可与多数的β-内酰胺酶牢固结合,生成不可逆的结合物,具有强力而广谱的抑制β-内酰胺酶作用。
阿莫西林制剂配合克拉维酸钾使用时能有效稳定阿莫西林,使其降解减少而疗效增强,且对革兰阳性及革兰阴性细菌均有效[1-3]。
有关物质是药物中主要疗效成分在储存中产生或在原料制备中引进的,说明了药物对温度、湿度、光等外在因素的稳定度及药物本身使用中的安全性。
含量均匀度是药物有效成分含量较小和比例较低时,在制剂的制备中可能引起分布不均匀而影响药物的质量。
药分试验四—制剂含量均匀度的检查概要

半固体制剂以及非均相液体制剂的每片(个)含
量符合标示量的程度,即每片(个)含量偏离标示量
的程度。
因待测样品比较多,因此一般选用简单易行、灵
敏度高的紫外分光光度法来测定药物的含量均匀 度。
重量差异试验
普通片剂 (混合均匀) 片重差异
含量均匀度
小剂量片剂或较难混合 均匀者 每片含量偏离标示量的 程度
不符合规定
含量均匀度检查法的判别线和复试线
③ A+S>15.0 不符合规定 ② A+1.80S>15.0 且A+S≤15.0 符合规定
S
15.0
复试区
10.0
5.0
①A+1.80S≤15.0 符合规定
5.0 10.0 15.0算
A ECL
A
c l c l A
适用范围
检查指标
特点
简便快速
准确
需要检查含量均匀度的制剂
除另有规定外,
片剂、胶囊剂或注射用无菌粉末,每片(个)标示量小
于25mg或主药含量小于每片(个)重量25%者;
内容物非均一溶液的软胶囊、单剂量包装的口服混悬液、 透皮贴剂、吸人剂和栓剂; 药物的有效浓度与毒副反应浓度比较接近的品种或混匀 工艺较困难的品种,每片(个)标示量不大于25mg;
孔滤膜( 0.45μm )滤过,取续滤液照紫外 - 可见分光光
度法(附录Ⅳ A)在261nm的波长处测定吸收度。限度为
±20%,应符合规定(附录ⅹ E)。
制剂含量均匀度检查一般步骤
取供试品 10 片, 分别测定每片以 标 示 量 为 100 的
求所得X的均值 及标准差S
A 100 X
中药制剂含量均匀度的测定

中药制剂含量均匀度的测定摘要:目的:以自制的银黄片为材料,分析高效液相色谱检测含量均匀度评价结果,分析采用红外线光谱分析最小二乘法检测不同处理方法的含量预测效果。
方法:把金银花提取物、黄芩提取物、预胶化淀粉和硬脂酸镁按照一定的比例进行等量递增混合,压制成片。
对药品进行溶解,分别利用高效液相色谱法和最小二乘法红外线光谱对药物的成分进行检测。
结果:不同红外线光谱处理分析法不同成分含量预测误差均方根多组之间的对比,差异有统计学意义(P<0.05)。
其中以经典MSC法获得的四种成分误差均方根值最低。
利用高效液相色谱法对四批金银花提取物检测的变异指数均在5%以内,具有较强的一致性。
结论:高效液相色谱是测定中药制剂含量均匀度的首选方法,检测精度高;红外线光谱分析具有一定的含量均匀度分析潜力,在控制误差的前提下能够较好的反映含量均匀度。
关键词:中药制剂;含量均匀度;测定[中图分类号]R286.0[文献标识吗]A [文章编号]1439-3768-(2019)-04-WJK 中药制剂稳定性研究是中药新药研究与开发的一项重要内容,是保证中药制剂有效性和安全性的重要基础。
制剂若发生分解、变质,不仅会影响其外观,而且可引起有效成分的含量变化和临床疗效的降低,导致药品失效,甚至产生或增加毒副作用,危及患者的健康和生命安全。
近年来中药因稳定性问题引起不良反应的报道逐年增多,给新药研发亮了“红灯”。
现阶段,尽管中药制剂稳定性研究得到普遍重视,但由于中药制剂成分的多样性,发生降解反应机制相当复杂,而且某些有效成分尚不明确,稳定性研究面临重重困难。
中药制剂含量测定是控制制剂质量的主要方法,是检验制剂稳定性关键,但现阶段稳定性研究含量测定仍存在不少漏洞,中药制剂稳定性考察急须处理好一些关键问题。
笔者总结了中药制剂稳定性研究概况,并针对制剂稳定性研究中含量测定的问题进行探讨,为稳定性研究工作规范化提供参考。
1材料与方法1.1实验仪器与材料本次实验材料包括金银花提取物、黄芩提取物、预胶化淀粉、乙腈以及超纯水等。
药品验证指南混合均匀度

药品验证指南混合均匀度英文回答:Drug Verification Guidelines for Homogeneity.Introduction:Ensuring the homogeneity of pharmaceutical products is crucial for their quality, safety, and efficacy. Homogeneity refers to the uniform distribution of active pharmaceutical ingredients (APIs) and other components within a drug product. This article aims to provide guidelines for evaluating the homogeneity of drugs.Methods for Homogeneity Testing:1. Visual Inspection:Visual inspection involves observing the drug product for any visible signs of non-uniformity, such asdiscoloration, clumping, or separation. This method provides a preliminary assessment of homogeneity but may not be sufficient for accurate quantification.2. Content Uniformity Testing:Content uniformity testing involves analyzing multiple samples from different locations within a batch to determine the consistency of API content. This can be done using various analytical techniques, such as high-performance liquid chromatography (HPLC) or spectrophotometry. Acceptance criteria for content uniformity are usually specified in pharmacopoeial monographs or regulatory guidelines.3. Blend Uniformity Testing:Blend uniformity testing is specifically applicable to drug products prepared by blending multiple components. It assesses the uniform distribution of APIs and excipients throughout the blend. Sampling methods, such as stratified sampling or systematic random sampling, should be employedto ensure representative samples are obtained.4. Statistical Analysis:Statistical analysis can be employed to evaluate the homogeneity of drug products quantitatively. Techniquessuch as analysis of variance (ANOVA) or chi-square test can be used to determine if there are significant differences among samples taken from different locations within a batch.Importance of Homogeneity Testing:Homogeneity testing is essential for several reasons:1. Ensuring Safety and Efficacy:Non-uniform distribution of APIs can lead toinconsistent drug potency, resulting in ineffective treatment or adverse effects. Homogeneity testing helps ensure that each dose contains the intended amount of API, minimizing the risk of under or over-dosing.2. Regulatory Compliance:Regulatory authorities require pharmaceutical manufacturers to demonstrate the homogeneity of their drug products. Compliance with these regulations is necessaryfor obtaining marketing approvals and maintaining product quality throughout the manufacturing process.3. Batch-to-Batch Consistency:Homogeneity testing also ensures consistency in drug quality and performance across different batches. This is particularly important for products with narrow therapeutic indices, where small variations in API distribution can significantly impact safety and efficacy.Conclusion:Homogeneity testing plays a critical role in pharmaceutical quality control. It helps identify and mitigate the risk of non-uniform distribution of APIs and other components within drug products. By following theguidelines mentioned above, manufacturers can ensure the safety, efficacy, and regulatory compliance of their pharmaceutical products.中文回答:药品混合均匀度验证指南。
含量均匀度检查法操作规程

含量均匀度检查法操作规程一、仪器和试剂准备1.高效液相色谱仪(HPLC)及其附件;2.所需的标准品和质量控制品;3.色谱柱:选择合适的色谱柱,确保其适应检测的活性成分;4.进样器:设定合适的进样体积,保证样品浓度适中;5.洗涤溶剂和流动相:按照方法要求配置好洗涤溶剂和流动相;6.离心机:用于制备样品溶液;7.干燥器:用于干燥样品或试剂;8.重量计:用于称取试剂和样品;9.pH计:用于测定溶液的pH值;10.紫外可见分光光度计:用于检测溶液的吸光度。
二、操作步骤1.样品的制备(1)将样品药品的数量根据检测要求称取到干燥器中;(2)打开干燥器,将药品在低温下进行干燥,直到其完全干燥;(3)关闭干燥器,等待样品冷却;(4)将干燥后的样品用适量的洗涤溶剂溶解,并进行适当的振荡和离心;(5)取出上清液,即为样品溶液。
2.仪器的配置(1)打开HPLC仪器的电源,并进行预热和平衡;(2)将色谱柱连接好,保证其正常的流动相;(3)调整进样器的体积,设定合适的样品进入量;(4)检查流速、温度和检测波长等参数的设置是否正确。
3.样品的进样(1)用吸管或进样器将样品溶液吸入色谱柱;(2)调整进样器的进样速度和体积,确保样品进入量合适。
4.HPLC方法运行(1)启动HPLC仪器,进入相应的方法运行界面;(2)设置合适的流动相速度,保证样品的快速流动;(3)检测样品的吸光度,记录峰高和峰面积等数据。
5.数据分析与结果记录(1)将样品的吸光度数据转化为样品中活性成分的含量;(2)对各个样品进行含量均匀度分析;(3)判断样品的含量均匀度是否合格;(4)记录各个样品的含量均匀度结果。
6.结果的解释与判断(1)根据含量均匀度检查结果,判断样品的质量是否合格;(2)当样品的含量均匀度达到要求时,判定为合格;(3)当样品的含量均匀度不达标时,判定为不合格;(4)根据不同样品的含量均匀度结果,进行相应的药品调整或重配。
三、操作注意事项1.严格按照方法要求进行操作;2.仔细准备和称取样品和试剂,避免误差;3.所有仪器和试剂都要按照要求进行校准和质量验证;4.样品制备时要避免污染和杂质的存在;5.检查样品进样的速度和体积,保证其准确性和一致性;6.在HPLC方法运行过程中,监测仪器和管路的状态,并及时处理异常情况;7.数据分析和结果判定要准确、客观、合理;8.结果的解释和判断要全面、客观,并根据相应的质量标准进行评价;9.操作结束后,及时清洗和维护仪器设备,确保其正常使用。
盐酸马普替林片含量均匀度检验不合格原因分析

盐酸马普替林片含量均匀度检验不合格原因分析作者:黄静杨东红来源:《中小企业管理与科技·下旬》2010年第09期摘要:盐酸马普替林片为选择性去甲肾上腺素再摄取抑制剂,是临床常见的抗抑郁药。
由于其为精神类药,且副作用以胆碱能阻断症状常见,如口干、便秘、视力模糊等,还可出现皮炎和皮疹,所以临床应严格控制其用量;且盐酸马普替林原料密度较制剂过程中使用的其他辅料的密度小,所以混匀工艺较困难:按照中国药典2005年版二部附录XE的规定:对于药物有效浓度与毒副反应浓度比较接近的品种或混匀工艺较困难的品种,每片(个)标示量不大于25mg者,应检查含量均匀度。
含量均匀度检查法以统计学理论为指导,综合标准差与偏离度而拟定的计量型方法。
关键词:盐酸马普替林片含量均匀度0 引言由于盐酸马普替林片含量均匀度测定方法较复杂,操作过程中影响因素较多,极易造成误判的风险:即本身合格的药品由于操作误差导致结果标准差和偏离度都很大,判为不合格品。
1 材料与方法试剂与样品:0.05mol/L盐酸溶液(多多药业) 磷酸盐缓冲液(PH3.5)(多多药业) 溴甲酚绿溶液(多多药业) 三氯甲烷(国药集团化学试剂有限公司) 盐酸马普替林片2 分析方法2.1含量均匀度测定法:随机取本品10片,分置100ml量瓶中,加0.05mol/L盐酸溶液约60ml,振摇使盐酸马普替林溶解,用0.05mol/L盐酸溶液稀释至刻度,摇匀,滤过,精密量取续滤液3ml,置100ml量瓶中,用磷酸盐缓冲液(pH3.5)(取磷酸二氢钾13.61g,加水适量使溶解,加0.1mol/L盐酸溶液50ml,加水稀释至2000ml)稀释至刻度,摇匀,作为供试品溶液;另取盐酸马普替林对照品25mg,精密称定,置100ml量瓶中,用0.05mol/L盐酸溶液溶解并稀释至刻度,摇匀;精密量取3ml,置100ml量瓶中,用磷酸盐缓冲液(pH3.5)稀释至刻度,摇匀,作为对照品溶液。
含量均匀度检查法
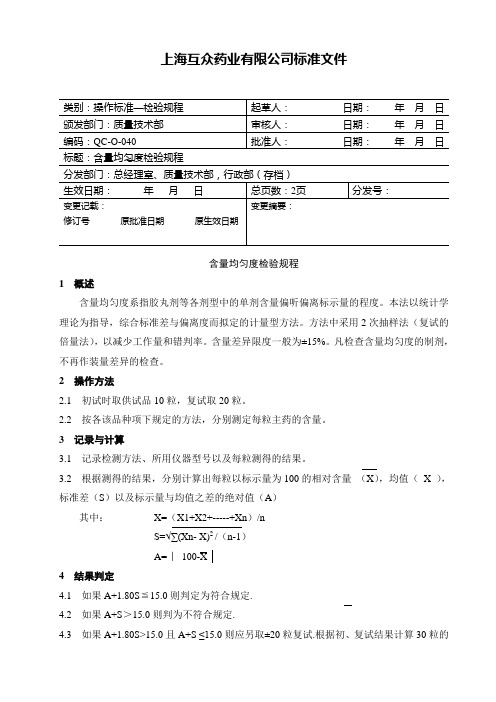
上海互众药业有限公司标准文件标题:含量均匀度检验规程含量均匀度检验规程1 概述含量均匀度系指胶丸剂等各剂型中的单剂含量偏听偏离标示量的程度。
本法以统计学理论为指导,综合标准差与偏离度而拟定的计量型方法。
方法中采用2次抽样法(复试的倍量法),以减少工作量和错判率。
含量差异限度一般为±15%。
凡检查含量均匀度的制剂,不再作装量差异的检查。
2 操作方法2.1 初试时取供试品10粒,复试取20粒。
2.2 按各该品种项下规定的方法,分别测定每粒主药的含量。
3 记录与计算3.1 记录检测方法、所用仪器型号以及每粒测得的结果。
3.2 根据测得的结果,分别计算出每粒以标示量为100的相对含量(X),均值(X ),标准差(S)以及标示量与均值之差的绝对值(A)其中:X=(X1+X2+-----+Xn)/nS=√∑(Xn- X)2 /(n-1)A= ︳100-X4 结果判定4.1 如果A+1.80S≦15.0则判定为符合规定.4.2 如果A+S>15.0则判为不符合规定.4.3 如果A+1.80S>15.0且A+S ≤15.0则应另取±20粒复试.根据初、复试结果计算30粒的2/2 含量均匀度检验规程QC-O-040 X、S、A。
若A+1。
45S≤15.0,即判为符合规定,如A+1.45S>15.0则判为不符合规定.4.4 如该品种项下规定含量均匀度的限度为20%或其他百分数时,应将上述各述各判定式中的15.0改为20.0或其他相应的数值,但各判定式中的系数不变.5 注意事项5.1 供试品的主药必须溶解完全,并定量转移至容量瓶中.5.2 测定所用的溶剂需一次配够,当用量较大时,即使是同批号的溶剂,也应混合均匀后使用。
含量均匀度检查

2、适用范围
除另有规定外,片剂、胶囊或注射用无菌粉末,每片(个)标 示量不大于25ml或主药含量不大于每片(个)重量25%者;小于 10mg或主药含量小于每片个重量的5mg;其它制剂,如内容物非均 一溶液的软胶囊、单剂量包装的口服混悬液、透皮贴剂、吸入剂和 栓剂,均应检查含量均匀度。 复方制剂仅检查符合上述条件的组分。
4、检查方法
除另有规定外,取供试品10片(个),按各种药品项下规定的方 法进行。 (1)分别测定每片(个)以标示量为100的相对含量X; (2)求10片(个)相对含量的平均值 x、标准差S、标示量与平均 值之差的绝对值A。
5、结果判定
例:地西泮
6、注意事项
(1)凡检查含量均匀度的制剂,不用检查重(装)量的差异。 (2)供试品的主药必须溶解完全,必要时可用乳钵研磨或超声处理, 促使溶解,并定量转移至容量瓶中。 (3)测定时溶液必须澄清,过滤不清,可离心后,取澄清液测定。 (4)即使是同批号的溶剂,也应混合均匀后使用。
3、必要性
(1)含量均匀度是考察制剂工艺水平的重要指标之一。以前是用装 (重)量差异来衡量的。但随着科学技术的发展及临床要求,装(重) 量差异检查不能满足对小剂量制剂质量控制的要求。重(装)量差异 是检查主药和辅料的总限度,没有明确考察主药的均一性,而主药是 起治疗作用的,尤其是对那些治疗剂量与中剂量比较接近的品种,控 制其含量均匀度显得尤为重要。匀度检查
四组
目
01
概念
2 0 1 7
录
02
适用范围
CONTENTS
03
必要性
04
检查方法
05
结果判定
06
注意事项
1、概念
含量均匀度是指小剂量 或单剂量的固体制剂、半 固体制剂和非均相液体制 剂的每片(个)含量符合 标示量的程度。 目的:控制每片(个) 含量的均一性,以保证用 药剂量准确。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
德信诚培训网
更多免费资料下载请进: 好好学习社区
药品试液配制含量均匀度检查法
一 目 的:制定试液的配制方法,规范试液的配制,保证测定结果的准确。
二 适用范围:适用于试液的配制。
三 责 任 者:品控部。
四 正 文:
1 简述
1.1 本法适用于中国兽药典2005年版一部附录(101页)含量均匀度检查。
1.2 含量均匀度系指粉剂或预混剂含量偏离标示量的程度。
1.3 除另有规定外主药含量小于每个重量2%者,均应检查含量均匀度。
复方制剂权检查符合上述条件的组分。
1.4 凡检查含量均匀度的制剂,不再测定含量,可用此平均含量结果作为含量测定结果。
1.5 本法以统计学理论为指导,综合标准差与偏离度而拟定的计量型方法。
含量均匀度的限度一般为±15%。
2 仪器与用具
按正文中该品种项下的规定。
3 试药与试液
按正文中该品种项下的规定。
4 操作方法
4.1 供试品10个。
4.2 取供试品,按照各该品种项下规定的方法,分别测定每片(个)主药的含量响应值。