强直性肌营养不良诊疗指南【2019版】
《罕见病诊疗指南(2019年版)》要点 (1)

《罕见病诊疗指南(2019年版)》要点(1)总目录1.21-羟化酶缺乏症2.白化病3.奥尔波特综合征4.肌萎缩侧索硬化5.天使综合征6.精氨酸酶缺乏症7.窒息性胸腔失养症(热纳综合征)8.非典型溶血性尿毒症综合征9.自身免疫性脑炎10.自身免疫性垂体炎11.自身免疫性胰岛素受体病12.β-酮硫解酶缺乏症13.生物素酶缺乏症14.心脏离子通道病15.原发性肉碱缺乏症16.Castleman病17.腓骨肌萎缩症18.瓜氨酸血症19.先天性肾上腺发育不良20.先天性高胰岛素性低血糖血症21.先天性肌无力综合征22.先天性肌强直23.先天性脊柱侧凸24.冠状动脉扩张25.先天性纯红细胞再生障碍性贫血26.Erdheim-Chester 病27.法布里病28.家族性地中海热29.范科尼贫血30.半乳糖血症31.戈谢病32.全身型重症肌无力33.Gitelman 综合征34.戊二酸血症型35.糖原累积病(型、型)36.血友病37.肝豆状核变性38.遗传性血管性水肿//39.遗传性大疱性表皮松解症40.遗传性果糖不耐受症41.遗传性低镁血症42.遗传性多发脑梗死性痴呆43.遗传性痉挛性截瘫44.全羧化酶合成酶缺乏症45.高同型半胱氨酸血症46.纯合子家族性高胆固醇血症47.亨廷顿病48.HHH 综合征(高鸟氨酸血症-高氨血症-同型瓜氨酸尿症)49.高苯丙氨酸血症50.低磷酸酯酶症51.低血磷性佝偻病52.特发性心肌病53.特发性低促性腺激素性性腺功能减退症54.特发性肺动脉高压55.特发性肺纤维化56.IgG4 相关性疾病57.先天性胆汁酸合成障碍58.异戊酸血症59.卡尔曼综合征60.朗格汉斯细胞组织细胞增生症61.莱伦综合征62.Leber 遗传性视神经病变63.长链-3-羟酰基辅酶A 脱氢酶缺乏症64.淋巴管肌瘤病65.赖氨酸尿蛋白不耐受症66.溶酶体酸性脂肪酶缺乏症67.枫糖尿症68.马方综合征69.McCune-Albright 综合征70.中链酰基辅酶A 脱氢酶缺乏症71.甲基丙二酸血症72.线粒体脑肌病73.黏多糖贮积症74.多灶性运动神经病75.多种酰基辅酶A 脱氢酶缺乏症76.多发性硬化77.多系统萎缩78.强直性肌营养不良79.N-乙酰谷氨酸合成酶缺乏症80.新生儿糖尿病81.视神经脊髓炎82.尼曼匹克病83.非综合征型耳聋84.努南综合征85.鸟氨酸氨甲酰基转移酶缺乏症86.成骨不全症87.帕金森病(青年型、早发型)88.阵发性睡眠性血红蛋白尿症89.波伊茨-耶格综合征90.苯丙酮尿症91.POEMS 综合征92.卟啉病93.普拉德-威利综合征94.原发性联合免疫缺陷病95.原发性遗传性肌张力不全96.原发性轻链型淀粉样变97.进行性家族性肝内胆汁淤积症98.进行性肌营养不良99.丙酸血症100.肺泡蛋白沉积症101.囊性纤维化102.视网膜色素变性103.视网膜母细胞瘤104.重症先天性粒细胞缺乏症105.婴儿严重肌阵挛性癫痫106.镰刀型细胞贫血病107.拉塞尔-西尔弗综合征Silver-Russell syndrome108.谷固醇血症109.脊髓延髓肌萎缩症(肯尼迪病) 110.脊髓性肌萎缩症111.脊髓小脑性共济失调112.系统性硬化症113.四氢生物喋呤缺乏症114.结节性硬化症115.酪氨酸血症116.极长链酰基辅酶A脱氢酶缺乏症117.威廉姆斯综合征118.湿疹-血小板减少-免疫缺陷综合征119.X连锁肾上腺脑白质营养不良120.X连锁无丙种球蛋白血症121.X连锁淋巴增生症资料(1)资料(1)目录1.21-羟化酶缺乏症2.白化病3.奥尔波特综合征4.肌萎缩侧索硬化5.天使综合征6.精氨酸酶缺乏症7.窒息性胸腔失养症(热纳综合征)8.非典型溶血性尿毒症综合征9.自身免疫性脑炎10.自身免疫性垂体炎1. 【21-羟化酶缺乏症】概述21-羟化酶缺乏症(21-OHD)是先天性肾上腺增生症(CAH)中最常见的类型,是由于编码21-羟化酶的CYP21A2 基因缺陷导致肾上腺皮质类固醇激素合成障碍的一种先天性疾病,呈常染色体隐性遗传。
肌营养不良

发病率:男性 多于女性
贝克氏肌营养不良症
01
病因:基 因突变
02
症状:肌 肉无力、 萎缩、行 走困难
03
发病率: 男性多于 女性
04
治疗方法: 药物治疗、 康复治疗、 基因治疗
其他类型肌营养不良症
面肩肱型肌 1 营养不良症
肢带型肌营 2 养不良症
远端型肌营 3 养不良症
眼肌型肌营 4 养不良症
强直性肌营 5 养不良症疾病发Leabharlann 过程123
4
初期:肌肉无力、 萎缩,运动功能减
退
中期:肌肉萎缩加 重,运动功能进一
步减退
终末期:呼吸肌萎 后期:肌肉萎缩严
缩,呼吸困难,危 重,运动功能丧失,
及生命
生活不能自理
杜兴氏肌营养不良症
症状:肌肉萎缩、 无力、行走困难
治疗方法:药物 治疗、康复治疗、
基因治疗等
病因:基因突 变导致
02
建立良好的人际关系,寻求家人和朋友的支持
03
学会自我调节,保持心理平衡
04
参加心理辅导和康复课程,提高心理素质
基因治疗
基因编辑技术: CRISPR/Cas
9等
基因治疗策略: 基因添加、基 因沉默、基因
编辑等
研究进展:动 物实验、临床
试验等
挑战与展望: 安全性、有效 性、伦理问题
等
干细胞治疗
干细胞来源:胚胎干细 胞、诱导多能干细胞等
治疗方法:干细胞移植、 基因编辑等
挑战与展望:安全性、 有效性、伦理问题等
干细胞类型:间充质干 细胞、神经干细胞等
研究进展:动物实验、 临床试验等
药物研发
基因治疗:通过基因编辑技术, 修复基因缺陷,改善症状
进行性肌营养不良诊疗指南【2019版】
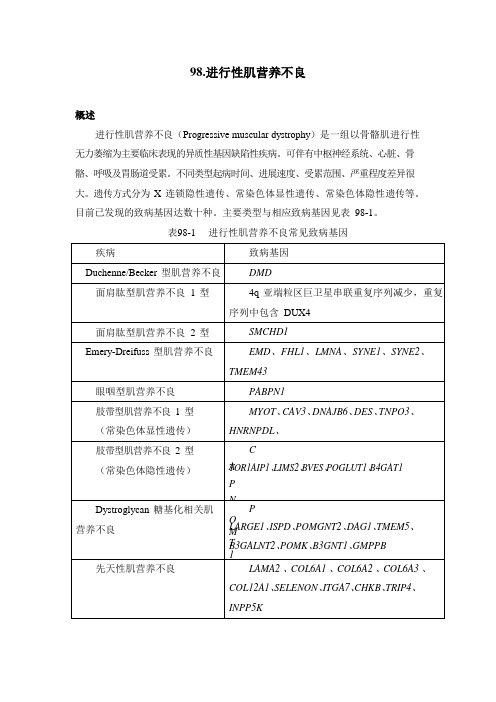
98.进行性肌营养不良概述进行性肌营养不良(Progressive muscular dystrophy)是一组以骨骼肌进行性无力萎缩为主要临床表现的异质性基因缺陷性疾病。
可伴有中枢神经系统、心脏、骨骼、呼吸及胃肠道受累。
不同类型起病时间、进展速度、受累范围、严重程度差异很大。
遗传方式分为X 连锁隐性遗传、常染色体显性遗传、常染色体隐性遗传等。
目前已发现的致病基因达数十种。
主要类型与相应致病基因见表98-1。
表98-1 进行性肌营养不良常见致病基因本组疾病虽有一定共性,但不同疾病诊治原则有很大不同。
下面以代表性疾病Duchenne/Becker 型肌营养不良(DMD/BMD)介绍相关诊疗常规。
病因和流行病学Duchenne/Becker 型肌营养不良的病因是维持肌肉细胞在伸缩过程中保持肌膜完整性的重要结构蛋白Dystrophin 的编码基因(DMD 基因)发生致病缺陷,从而导致功能异常,最终造成肌肉进行性破坏。
Duchenne/Becker 型肌营养不良遗传方式为X 连锁隐性遗传,发病率在各个国家、地区和人种间无明显差异,每3600~6000 出生男婴中有1 例发病。
我国的发病率约为1/3853,估算全国患者约70 000 人。
临床表现Duchenne 型肌营养不良在儿童期起病。
表现为运动发育轻度迟滞,骨骼肌进行性无力萎缩,影响肢体运动功能,逐渐出现步态异常、上肢活动受限,自然病程常在10 岁左右丧失行走能力。
此后出现脊柱侧弯、关节挛缩、呼吸肌无力、扩张性心肌病,20 岁左右因呼吸衰竭、心功能衰竭死亡。
查体可见双腓肠肌假肥大,同时可有双前臂及舌肌假肥大,Gower’s 征阳性,腰椎前凸等。
Becker 型肌营养不良为同一疾病的相对良性表型,因DMD 基因功能未完全丧失,所以病情明显轻于Duchenne 型肌营养不良。
可青年甚至成年起病,部分患者不影响生存期。
假肥大体征明显,部分患者在肢体无力尚轻时,先出现明显的扩张性心肌病。
儿童营养不良的诊疗指南

肌张力:肌张力的异常可能提示神经系统功能障碍。
运动发育:运动发育的延迟可能提示营养不良。
营养相关生化指标:如白蛋白、前白蛋白等,这些指标可 以反映近期营养状况。
诊断流程与步骤
01
02
03
04
05
1. 收集病史
2. 身体检查
3. 评估生长曲线 4. 检查是否有并 5. 实验室检查 发症
06
总结与展望
总结经验教训
营养不良是儿童健康的主要问题之一, 对儿童的生长、发育和认知能力都有负
面影响。
营养不良往往与贫困、不卫生和不良饮 食习惯等密切相关,因此需要从多方面
进行干预和治疗。
针对不同类型的营养不良,需要采取不 同的诊疗方法和策略,例如针对蛋白质 -能量营养不良需要进行能量和蛋白质 补充,针对微量营养素缺乏需要进行特
营养不良会影响儿童的脑部发 育,导致智力水平低下。
心理问题
长期营养不良可能导致儿童出 现自卑、抑郁等心理问题,影
响其心理健康。
02
儿童营养不良的诊断
诊断标准与方法
体重低于同年龄、同性别参照人群值的百分比:此标准适 用于年龄别体重和年龄别身高评价。
生长曲线:与生长曲线进行比较,观察其是否在正常范围 内。
分类
根据病因,儿童营养不良可分为原发性营养不良和继发性营 养不良。原发性营养不良主要是由于摄入不足、吸收不良或 过度消耗导致,继发性营养不良则是由疾病或药物影响导致 。
营养不良的原因
01
02
03
04
饮食不当
儿童饮食单一、食物质量差、 摄入不足或偏食等都可能导致
营养不良。
疾病影响
强直性肌营养不良症护理查房

强直性肌营养不良症护理查房
1. 介绍
强直性肌营养不良症(SMA)是一种罕见的遗传性疾病,主要影响神经系统,导
致肌肉无法正常发育和运动功能丧失。
在SMA患者中,不同的类型和程度的病情需
要不同的护理措施。
2. 查房内容
在对SMA患者进行护理查房时,护士应该注意以下几个方面:
2.1 生命体征监测
定期监测患者的生命体征是护理查房的基础。
重点关注呼吸、心率、血压等指标,及时发现异常情况并采取相应措施。
2.2 营养支持
SMA患者的肌肉功能受损,容易出现吞咽困难等问题,护士应确保患者获得足够的营养支持。
定期评估患者的饮食情况,必要时进行营养补充。
2.3 皮肤护理
长期卧床的SMA患者容易出现压疮等皮肤问题,护士要定期为患者翻身、按摩,保持皮肤清洁,防止皮肤损伤。
2.4 康复训练
康复训练对于SMA患者的康复非常重要。
护士要协助患者进行康复训练,维持肌肉功能,帮助患者尽可能恢复或保持活动能力。
2.5 心理支持
SMA患者常常面临生活的困难和挑战,护士要给予患者必要的心理支持,帮助他们建立积极的心态,面对疾病。
3. 结束语
通过对SMA患者的护理查房,护士可以及时了解患者的病情和护理需求,为患者提供全面的护理服务,促进患者的康复和生活质量的提高。
希望护士们能够认真负责地执行护理查房工作,为SMA患者提供更好的护理服务。
肌营养不良症的科普知识PPT课件

什么是肌营养不良症?
类型
肌营养不良症有多种类型,最常见的是杜氏肌营 养不良症和贝克肌营养不良症。
不同类型的肌营养不良症在症状和发病年龄上有 所不同。
什么是肌营养不良症?
发病机制
肌营养不良症的发病机制通常涉及肌养不良症?
新兴治疗
基因疗法和干细胞疗法等新兴治疗方法正在 研究中,有望为患者带来新的希望。
临床试验正在进行中,患者可以咨询医生了 解更多信息。
如何治疗肌营养不良症?
生活方式干预
保持适度的运动和良好的营养可以帮助患者 更好地管理症状。
定期的身体锻炼可以增强肌肉力量和耐力, 改善生活质量。
肌营养不良症的症状是什么?
肌营养不良症的症状是什么?
主要症状
肌肉无力、肌肉萎缩、运动能力下降是肌营养不 良症的主要症状。
患者可能会出现走路困难、爬楼梯时感到吃力等 情况。
肌营养不良症的症状是什么? 其他表现
部分患者可能伴随心脏和呼吸系统的问题,尤其 在疾病晚期。
心肌病是某些类型肌营养不良症的常见并发症。
例如,杜氏肌营养不良症涉及到肌肉细胞膜的一 个重要蛋白质——德兴蛋白的缺失。
谁会得肌营养不良症?
谁会得肌营养不良症?
遗传因素
肌营养不良症通常是由遗传因素引起的,许 多类型以X连锁隐性遗传方式传递。
这意味着男性更容易受到影响,因为他们只 有一个X染色体,而女性则有两个。
谁会得肌营养不良症?
发病年龄
肌营养不良症的症状是什么?
早期诊断
早期识别症状并进行检查至关重要,以便及时采 取治疗措施。
常见的检查方法包括肌电图、肌肉活检以及基因 检测等。
糖原累积病(Ⅰ型和Ⅱ型)诊疗指南(2019)

Chinese Practical Journal of Rural Doctor 2021 Vol.28 No.3标准•方案•指南糖原累积病(I型和I I型)诊疗指南(2019)中华人民共和国国家卫生健康委员会1概述糖原累积病(I型、u型)均是常染色体隐性 遗传病。
GSD I a型是由于G6PC突变使肝脏葡萄 糖-6-磷酸酶缺乏所致。
典型表现为婴幼儿期起病 的肝脏肿大、生长发育落后、空腹低血糖、高脂血 症、高尿酸血症和高乳酸血症等。
GSD I b型是由 于SLC37A4基因突变使葡萄糖-6-磷酸转移酶缺 乏所致。
患者除了有I a型表现之外,还可有粒细 胞减少和功能缺陷的表现。
GSD II型是由GAA突 变导致a-1,4-葡萄糖苷酶缺陷,造成糖原堆积在 溶酶体和胞质中,使心肌、骨骼肌等脏器损害。
根 据发病年龄、受累器官、严重程度和病情进展情 况可分为婴儿型((infantile-onset pompe disease, IOPD)和晚发型(late-onset pompe disease,LOPD)。
2病因和流行病学GSD I a型致病基因G6PC位干17q21,含5个 外显子,基因突变导致糖原降解或异生过程不能 释放葡萄糖,使6-磷酸葡萄糖堆积,通过糖酵解途 径产生过多乳酸,通过磷酸戊糖途径致血尿酸升 高,同时生成大量乙酰辅酶A,致血脂升高。
至今 已报道的G6PC突变达116种,中国人最常见突变 是 c.648G>T(56.3%~57%)和 c_248G>A(12.1% ~14%)。
080I b型致病基因SLC37A4位于llq23, 含9个外显子,基因产物为跨膜蛋白葡萄糖-6-磷 酸转移酶,其作用是将葡萄糖-6-磷酸从细胞浆和 内质网膜间隙转运到内质网腔内。
当基因突变导 致葡萄糖-6-磷酸转移酶缺乏时,葡萄糖-6-磷酸 不能被转运到微粒体膜而进一步水解产生葡萄 糖,造成与糖原累积症I a型相同的表现。
强直性肌营养不良症40页PPT

1.手及前臂伸肌的萎缩,也可能以眼脸 下垂及面肌松弛为最早出现的表现。 常见有胸锁乳突肌无力及变薄,呈现 似“鹅颈”。
2.咽、喉无力表现为单音调及鼻音。横 膈无力导致低通气。心脏传导异常, 表现为心率慢,P-R延长。
3.肌强直现象。表现为主动收缩时放 松延慢,及叩诊性肌强直。易在前 臂、手及舌引出。肌强直可于萎缩 前数年即出现。
4.非肌肉组织的营养不良改变,以晶 体透明度减低最为常见。
5.其他如:前额秃发,咀嚼肌萎缩因 而呈现似斧状脸等。
肌强直现象
叩诊性肌强直
肌电图所见
谢谢
11、越是没有本领的就越加自命不凡。——邓拓 12、越是无能的人,越喜欢挑剔别人的错儿。——爱尔兰 13、知人者智,自知者明。胜人者有力,自胜者强。——老子 14、意志坚强的人能把世界放在手中像泥块一样任意揉捏。——歌德 15、最具挑战性的挑战莫过于提升自我。——迈克尔·F·斯特利
强直性肌营养不良症
强直性肌营养不良
Myotonic muscular dystrophy Dystrophia Myotonica Myotonia Atrophica
Curschmann-Steinert Disease
缺陷基因位于19q13.3
三核苷CTG(胞嘧啶、胸腺嘧啶及 胍)序列比正常人为大。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
77. 强直性肌营养不良
概述
强直性肌营养不良(myotonic dystrophy ,DM )是以肌强直现象(主动或被动肌肉收缩后无法及时放松)和肌肉进行性无力萎缩为主要特点的进行性肌营养不良类型。
除肌肉受累外,强直性肌营养不良累及全身多个器官系统,包括眼、心脏、内分泌系统和中枢神经系统。
强直性肌营养不良分为 1 型和 2 型,1 型 (DM1)由 DMPK 基因 3’端非编码区 CTG 三核苷酸重复序列异常增多所致,2 型(DM2)由 ZNF9(CNBP )基因 1 号内含子 CCTG 四核苷酸重复序列异常增多所致。
1 型较为常见,2 型相对少见且病情较轻,患者间差异较小。
病因和流行病学
强直性肌营养不良 1 型、2 型的异常增多重复序列均位于非编码区。
目前认为发病机制为异常 RNA 毒性理论。
当重复序列异常增多后,在基因转录为 RNA 后,不再进一步翻译为蛋白质,而是形成发卡结构留存在细胞核内。
这些异常 R
N A 占
据了与
剪切
密
切相关
的重要
R N
A
结
合
蛋
白
家
族和
CUG-BP and ETR-3-like-factors (CELF ),使其不能行使正常功能,从而引起多种下游蛋白剪切异常、功能受损,最终造成多系统受累表现。
强直性肌营养不良是成年起病的最常见肌营养不良类型。
估计患病率约为1/8000。
D M1 较DM2 多见,但也有部分国家流行病学调查显示 DM2 患者与DM1 患者相当,甚至更多。
临床表现
强直性肌营养不良因 RNA 毒性作用导致多系统受累,主要临床表现如下: 1. 肌肉 四肢远端起始并逐渐向近端进展的无力萎缩,影响手部精细动作, 造成垂足。
面肌亦可受累。
同时有明显肌强直现象,累及肢体肌、咽喉肌等。
查体可发现大力握拳后手指不能立即展开,用力闭目后不能立即睁开。
叩击手部大鱼际肌、舌肌等,可引出肌强直,称为叩击性肌强直或“肌球”现象。
肌强直存在“热身”现象,反复活动后,肌强直减轻。
下肢肌痛和全身疲乏感也是常见临
床表现。
2.心脏心脏传导系统常有受累,出现不同程度的房室传导阻滞和室内传导异常。
严重时可造成猝死。
确诊患者需定期监测心电图,必要时植入起搏器。
3.中枢神经系统睡眠增多、阻塞性睡眠呼吸暂停等睡眠障碍多见。
可伴有认知及情绪障碍。
头M RI 可见弥漫性脑白质病变及皮层萎缩。
4.眼随着年龄增长,几乎所有患者均会出现白内障。
常在30~40 岁影响视力。
5.内分泌系统可出现多种内分泌疾病,包括糖尿病、甲状腺功能异常、钙磷代谢异常、性激素异常、睾丸萎缩(造成不育)等。
此外,还可有额秃、胃肠道动力不足、限制性通气障碍等。
DM1 型随着CTG 重复数增多,病情逐渐加重。
一般从轻到重分为轻型、经典成人型、儿童型、先天型。
在同一家族中常呈现“遗传早现”,即子代发病更早、症状更重。
DM2 临床表现与DM1 类似,通常程度较轻,婴幼儿起病或重症患者罕见,无“遗传早现”。
颈肌和肢体近端肌肉受累更早、更明显。
辅助检查
1.肌电图针极肌电图可见肌强直放电,同时合并肌源性损害电生理表现,对诊断具有重要意义。
2.血清肌酶谱检测肌酸激酶(CK)可正常或轻度升高,可达正常上限的3~
4 倍。
3.肌肉活检病程不同时期的肌肉病理改变有很大差异,早期可仅见肌纤维大小不等。
典型的肌肉病理改变包括中央核明显增多、大量肌纤维含有肌浆块、Ⅰ型纤维优势及萎缩。
目前根据临床特点、肌肉电生理改变和基因检测基本可以明确诊断,肌肉活检并非必要。
4.心电图或Holter 疑诊患者需行心电检测,明确有无房室传导阻滞或其他类型传导异常。
确诊后也需定期随诊心电图,如出现逐渐进展的心律失常,如Ⅲ 度房室传导阻滞,需考虑起搏器植入。
5.眼科检查通过眼科裂隙灯等检查,明确有无白内障,并予针对性治疗。
6.内分泌检测对血糖、甲功、钙磷代谢、性激素等方面做全面评估,明确有无内分泌异常,并予相应随诊治疗。
7.其他方面如有认知、情绪、睡眠等方面问题,应行头MR、睡眠监测、认知及情绪量表评估等,并予对症处理。
8.基因检测根据临床特点判断分型,然后分别对致病基因DMPK、ZNF9 进行检测。
因重复数可能高达数千,对重复序列的检测一般采用特殊的TP-PCR 方法,重复数特别大时,可采用Southern blot 法检测。
诊断
强直性肌营养不良的诊断依靠病史(常染色体显性遗传家族史、“遗传早现”现、特征性体征(远端或轴位为主肌肉无力萎缩、肌病面容、肌球象和肌强直现象)
、肌电图、心电图、眼科、内分泌检查发现多系统损害,最终通过现象、额秃等)
基因检测确诊。
鉴别诊断
强直性肌营养不良主要需要与其他强直性肌病或遗传性肌病相鉴别。
1.先天性肌强直(myotonia congenita)患者通常儿童早期起病,有明显肌强直现象,常有肌肉肥大,呈运动员体型,强直症状经反复运动热身后减轻。
病程更为良性,不伴进行性肌肉无力和肌萎缩。
肌电图检查可见肌强直放电,但无肌源性损害。
为CLCN1 基因缺陷所致。
2.先天性副肌强直(paramyotonia congenita)患者亦有肌强直现象,肌电图可见肌强直放电,但一般无肌源性损害。
症状以面肌、颈肌和上肢肌肉受累为
,遇冷时强直现象亦明显加重。
运主,在活动和反复动作后加重(无“热身”现象)
动员体型一般不明显。
患者常有发作性无力,持续数分钟至数小时。
为S CN4A 基因缺陷所致。
3.远端型肌病GNE 包涵体肌病、Miyoshi 肌病等远端型肌病肌肉无力萎缩特点与DM1 相似,但前两者没有典型肌强直现象,肌电图也没有典型肌强直放电以及缺少强直性肌营养不良多系统受累特点可资鉴别。
4.先天性肌病先天性DM1 患者病情严重,出生即表现为“松软儿”和呼。