番茄红素质量标准及检验规程
番茄红素的提取及其抗氧化能力的检测

番茄中番茄红素的提取及抗氧化活性评价摘要采用溶剂提取法从番茄中提取番茄红素,选用低毒性的乙酸乙酯进行提取,测定其提取率和纯度。
利用DPPH消除法对番茄红素与维生素E的抗氧化能力进行对比。
关键词番茄红素提取抗氧化检测1 前言番茄红素(lycopene)是一种脂溶性不饱和碳氢化合物,是胡萝卜素的一种,分子式C40H56,分子量536.85,熔点174℃。
其晶体呈红色,它在成熟的红色植物果实中,如番茄、西瓜、胡萝卜、草莓、柑橘中含量较高,由于最早从番茄中分离制得,故称番茄红素。
在流行病学研究中发现富含番茄红素的食物可以抑制多种癌症对人体的侵袭[1]。
其抗氧化作用和自由基消除作用被认为是其能抑制癌症发生的原因之一。
在减缓动脉硬化、预防心血管疾病等各种与衰老有关的疾病及增强机体免疫力方面也有重要的作用。
另外,番茄红素不仅可以作为保健食品,亦可用于化妆品,它可以防止紫外线、健肤养颜、延缓衰老[2]。
近年来国内外对番茄红素的研究方兴未艾,具有很高的经济价值和医学价值。
2 实验目的2.1掌握溶剂法从新鲜番茄中提取番茄红素。
2.2掌握检测番茄红素体外抗氧化性的检测方法。
3 实验原理3.1 番茄红素属于类胡萝卜素色素,与其他类胡萝卜素相比,它具有独特的长链分子结构,是一条两端开环的非极性碳氢链,为脂溶性物质,采用有机溶剂浸提法、超声辅助提取时最佳的提取溶剂是乙酸乙酯[3]。
提取温度50℃,总提取时间6min,间隔时间5s,超声波输出频率300w。
按料液比1:5浸提时可得最佳浸提效果[4]。
3.2以苏丹红代替番茄红素作为标准品,以含2%的氯仿为溶剂在518.8nm处测吸光度可以排除β胡萝卜素的干扰,提高分光光度法测定的准确性[5]。
3.3利用DPPH清除法分别检测番茄红素与维生素E的抗氧化能力,将二者抗氧化能力进行对比。
DPPH广泛用于定量测定生物试样、分类物质好坏和食品的抗氧化能力。
DPPH是一种很稳定的氮中心的自由基,它的稳定性主要来自共振稳定作用的3个苯环的空间障碍,使夹在中间的氮原子上不成对的电子不能发挥其应有的电子成对作用。
番茄红素实验报告

番茄红素实验报告1. 引言番茄是一种常见的蔬菜水果,富含多种营养物质,其中番茄红素是其主要色素成分之一。
番茄红素在许多植物和水果中都存在,具有抗氧化、抗癌等多种生理功能。
本实验旨在通过实验证明番茄中存在番茄红素,并通过分光光度法进行测定。
2. 实验原理番茄红素属于类胡萝卜素的一种,其分子结构中含有若干个双键,使其具有一定的共振特性,呈现出红色的吸光峰。
利用此特性,可以通过分光光度法对番茄红素进行测定。
分光光度法是通过测量溶液在特定波长下对光的吸收,来推测溶液中物质的浓度。
3. 实验步骤1. 将新鲜番茄取皮并剁碎,将番茄碎液放入砂浴加热器中加热20分钟,使其完全破裂释放番茄红素。
2. 过滤番茄碎液,去除残渣。
取得的番茄汁即为待测番茄红素溶液。
3. 使用分光光度计,设置波长为480nm,将番茄红素溶液置于比色皿中,放入分光光度计测量室,记录吸光度值。
4. 设立不同浓度的番茄红素标准溶液,并使用同样的方法测量其吸光度值,制作标准曲线。
5. 根据标准曲线,计算待测番茄红素溶液的浓度。
4. 实验数据标准溶液浓度(mg/L) 吸光度0.5 0.1201 0.2402 0.4904 1.0106 1.4708 2.10010 2.700待测番茄红素溶液的吸光度为0.930。
5. 结果与讨论通过测量待测番茄红素溶液的吸光度,并参考标准曲线,可以得出其浓度为3.244 mg/L。
因此,在所使用的实验条件下,我们成功地定量测定了番茄红素的浓度。
同时,通过对比标准溶液和待测溶液的吸光度,还可以推测番茄红素在不同浓度下的吸光度与浓度之间的线性关系。
然而,实验中可能存在一些误差。
首先,番茄红素的提取效率可能会受番茄的成熟度、存储方式等因素的影响。
其次,实验中的操作技巧和器材精确度也会对结果产生影响。
为了减小误差,可以多次重复实验并取平均值,同时使用优质的实验器材来提高数据的准确性。
6. 结论通过本实验,我们成功地分离出番茄红素并测定了其浓度为3.244 mg/L。
番茄红素含量报告

番茄红素含量报告引言番茄(学名:Solanum lycopersicum)是一种常见的蔬果,其红色的外皮和独特的味道深受人们喜爱。
番茄中含有丰富的番茄红素,这是一种天然的抗氧化剂,对于人体健康具有重要的意义。
本文将对番茄红素的含量进行测定和报告。
研究方法样品收集为了研究番茄红素的含量,我们选取了10个种类不同的番茄作为研究样本。
这些样本来自于当地的农田,并且在成熟的时候进行了采集。
样品处理在采集到番茄样本后,我们将其进行了处理以得到可测量的番茄红素。
处理方法如下:1.每个番茄样本被切成小块,并去掉番茄的皮;2.将切好的番茄块放入搅拌机中,加入适量的蒸馏水;3.打开搅拌机,将番茄块搅拌至均匀状态,制成番茄汁;4.将番茄汁过滤,去除固体残渣,得到纯净的番茄汁。
番茄红素含量的测定为了测定番茄红素的含量,我们使用了分光光度计。
测定步骤如下:1.将纯净的番茄汁分装到玻璃试管中,每管约装满2/3;2.将试管放入分光光度计中,设定波长为450nm;3.记录各样本的吸光度值;4.根据标准曲线,将吸光度值转换为番茄红素的浓度。
结果与讨论通过上述测定方法,我们得到了不同番茄样本中番茄红素的含量。
下表展示了10个样本的番茄红素含量(单位:mg/100g):样本编号番茄红素含量1 8.52 9.23 7.84 6.95 7.36 8.17 9.68 6.59 7.710 8.9从上表可以看出,不同番茄样本中番茄红素的含量有所差异,最低的样本为6.5mg/100g,最高的样本为9.6mg/100g。
这说明了番茄红素的含量与番茄的品种或生长环境有一定关系。
番茄红素被广泛认为是一种有效的抗氧化剂,可帮助人体抵抗自由基的损害。
它还被发现具有抗炎、抗癌和心血管保护等多种保健功效。
因此,食用富含番茄红素的番茄是维护健康的一种选择。
结论本研究报告对10个不同种类的番茄进行了番茄红素含量的测定。
结果表明,番茄红素的含量在不同番茄样本中有所差异。
番茄红素检测

番茄红素检测
番茄红素(Lycopene),又称为番茄素,是一种常见的类胡萝卜素,广泛存在于番茄、葡萄柑橘等果实中。
有的微生物也能合成番茄红素,如红色细菌。
番茄红素的抗氧化能力大约是β-胡萝卜素的3倍,可延缓衰老、预防癌症和心血管等疾病。
迪信泰检测平台采用高效液相色谱(HPLC)技术,使用Agilent或者Waters高效液
相色谱仪,配备DAD检测器或ELSD检测器,可检测各类样品中番茄红素含量变化。
此外,我们还提供其他多种色素检测服务,以满足您的不同需求。
送样要求和检测周期。
HPLC测定番茄红素样本要求:
1. 请确保样本量大于0.2g或者0.2mL。
周期:2~3周。
项目报告。
项目结束后迪信泰检测平台将会提供详细中英文双语技术报告,报告包括:
1. 实验步骤(中英文)。
2. 相关质谱参数(中英文)。
3. 质谱图片。
4. 原始数据。
5. 番茄红素含量信息。
迪信泰检测平台可根据需求定制其他物质测定方案,具体可免费咨询技术支持。
番茄红素(合成)技术指标检测标准
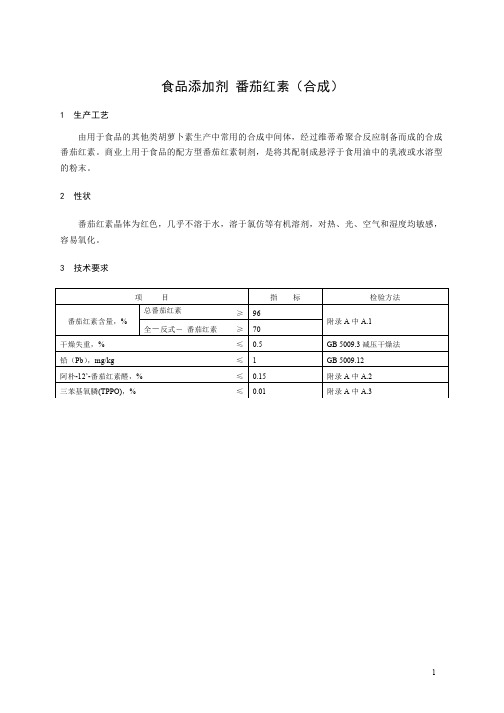
食品添加剂 番茄红素(合成)1 生产工艺由用于食品的其他类胡萝卜素生产中常用的合成中间体,经过维蒂希聚合反应制备而成的合成番茄红素。
商业上用于食品的配方型番茄红素制剂,是将其配制成悬浮于食用油中的乳液或水溶型的粉末。
2 性状番茄红素晶体为红色,几乎不溶于水,溶于氯仿等有机溶剂,对热、光、空气和湿度均敏感,容易氧化。
3 技术要求项目指标检验方法番茄红素含量,% 总番茄红素≥ 96附录A中A.1 全-反式-番茄红素≥ 70干燥失重,% ≤ 0.5 GB 5009.3减压干燥法铅(Pb),mg/kg ≤ 1 GB 5009.12阿朴-12’-番茄红素醛,% ≤ 0.15 附录A中A.2三苯基氧膦(TPPO),% ≤ 0.01 附录A中A.3附 录 A检验方法A.1 总番茄红素含量和全-反式-番茄红素的含量的测定A.1.1 方法原理用高效液相色谱法在下列条件下测定总番茄红素含量和全-反式-番茄红素的含量A.1.2 试剂和材料(注意:所有溶剂均应为色谱级)A.1.2.1 正己烷。
A.1.2.2 用0.025%特丁基对苯二酚(BHT)稳定的四氢呋喃。
A.1.2.3 n-乙基-二异丙胺。
A.1.2.4 番茄红素标准品纯度≥95%。
A.1.3 仪器和设备A.1.3.1紫外/可见分光光度计,配备1cm吸收池。
A.1.3.2 高效液相色谱系统,配备合适的泵、进样器、色谱柱恒温箱和积分仪。
A.1.3.3 色谱柱:2根相连的不锈钢柱(250x4.0 mm)。
A.1.3.4 固定相:Nucleosil 300-5,5µmA.1.3.5 HPLC条件流速:0.8 mL/min进样量:20 µL柱温:20℃检测波长:470 nm流动相:0.15% n-乙基-二异丙胺的正己烷溶液(V/V)A.1.3.6 HPLC测定用标准溶液:精密称取番茄红素标准品5.5-6.5mg,置100mL量瓶中,加入特丁基对苯二酚(BHT)稳定的四氢呋喃5ml使溶解,用正己烷稀释至刻度。
番茄红素国家标准

番茄红素国家标准引言番茄红素是一种天然色素,主要存在于番茄等植物中。
它具有抗氧化、抗炎和抗癌等多种保健功效,受到越来越多人的关注和研究。
为了保证番茄红素产品的质量和安全性,制定番茄红素国家标准是必不可少的。
本文将介绍番茄红素国家标准的制定背景、范围、主要内容以及实施和管理等方面内容。
制定背景随着人们对健康的关注度不断提高,番茄红素越来越受欢迎。
然而,市场上的番茄红素产品质量参差不齐,存在安全性和成分含量不合理等问题。
为了规范番茄红素产品的生产和销售,确保产品的质量和安全性,制定番茄红素国家标准成为必要措施。
范围番茄红素国家标准适用于番茄红素产品的生产、销售和检测等环节。
其中,番茄红素产品包括番茄红素提取物、番茄红素软胶囊等各种形式的产品。
主要内容番茄红素国家标准主要包括以下内容:1. 品质要求该部分规定了番茄红素产品的外观、质地、颜色、气味等方面的要求。
例如,番茄红素产品应具有红色至深红色的外观,并且没有异味。
2. 成分含量该部分规定了番茄红素产品中番茄红素的含量要求。
根据不同类型的番茄红素产品,规定了相应的番茄红素含量范围。
3. 残留物限量该部分规定了番茄红素产品中可能存在的农药残留、重金属等有害物质的限量要求。
这些限量值通常是根据食品安全标准和科学研究得出的。
4. 包装和标签该部分规定了番茄红素产品的包装和标签要求。
例如,番茄红素产品的标签应包括产品名称、生产日期、保质期、生产企业等信息,并且应采用明确、易识别的字体和颜色。
5. 检测方法该部分规定了番茄红素产品质量检测的方法和标准。
包括番茄红素含量的测定、残留物检测等方面的方法。
实施和管理番茄红素国家标准的实施和管理需要依靠相关部门的监督和执行。
在生产和销售环节,企业需要依据国家标准进行生产和管理。
同时,有关部门应加强对番茄红素产品的抽检和监督,确保标准的执行和产品质量的合格。
结论番茄红素国家标准的制定对于保障番茄红素产品的质量和安全性具有重要意义。
番茄红素软胶囊企业标准

Q/KSB009-2009 ICS 67.230标准检索号X83海南省企业标准Q/KSB009-2009番茄红素软胶囊2009-08-18发布 2009-08-18实施海南康斯宝生物技术有限公司发布Q/KSB009-2009番茄红素软胶囊1范围本标准规定了番茄红素软胶囊的技术要求、生产加工过程的卫生要求、试验方法、检验规则、标志、包装、运输和贮存。
本标准适用于以番茄红素、精制大豆油为主要原料,经溶胶、配料、压丸、干燥和包装而成的番茄红素软胶囊的生产控制、检验、销售等环节。
2规范性引用文件下列文件中的条款通过本标准的引用而成为本标准的条款。
凡是注日期的引用文件,其随后所有的修改单(不包括勘误表的内容)或修订版均不适用于本标准。
然而,鼓励根据本标准达成协议的各方研究是可使用这些文件的最新版本。
凡是不注日期的引用文件,其最新版本适用于本标准。
GB/T 191-2000 包装储运图示标志GB 1535 大豆油GB 14881 食品企业通用卫生规定GB/T 4789.2-2003 食品卫生微生物学检验菌落总数测定GB/T 4789.3-2003 食品卫生微生物学检验大肠菌群测定GB/T 4789.4-2003 食品卫生微生物学检验沙门氏菌检验GB/T 4789.5-2003 食品卫生微生物学检验志贺氏菌检验GB/T 4789.10-2003 食品卫生微生物学检验金黄色葡萄球菌检验GB/T 4789.11-2003 食品卫生微生物学检验溶血性链球菌检验GB/T 4789.15-2003 食品卫生微生物学检验霉菌和酵母计数GB/T 5009.4-2003 食品中灰分的测定GB/T 5009.11-2003 食品中总砷及无机砷的测定GB/T 5009.12-2003 食品中铅的测定GB/T 5009.17-2003 食品中总汞及有机汞的测定GB/T 5009.37-2003 食用植物油卫生标准的分析方法GB 6543-1986 瓦楞纸箱GB 6783-1994 食品添加剂明胶GB 7718-1994 食品标签通用标准GB/T 22249-2008 保健食品中番茄红素的测定JJF 1070 定量包装商品净含量计量检验规则GB 9687 食品包装用聚乙烯成型品卫生标准国家质量监督检验检疫总局令第102号《食品标识管理规定》《中华人民共和国药典》2005年版国家质量监督检验检疫总局(2005)第75号令《定量包装商品计量监督管理办法》3规格250mg/粒或450mg/粒或500mg/粒或800mg/粒或1000mg/粒,塑料瓶装或泡罩包装。
番茄酱检验规程

番茄酱检验规程1 范围本规程规定了番茄汁生产用原料番茄酱的技术要求,检验规则和包装运输的要求2 技术要求2.1感官指标:2.1.1组织性状及滋味:具有番茄酱应有的滋味和气味,无异味,色泽呈一致的深红色或红色,组织状态酱体均匀一致粘稠适度。
2.2.2杂质:无肉眼可见外来杂质。
2.2 理化要求2.3 微生物指标3.检验方法3.1感官指标:3.1.1组织性状及滋味:在常温下开袋,评定气味和滋味,将酱体倒入干净干燥的白瓷盘中,静置1min,观察其色泽、形态,有无流散和汁液分离现象。
3.1.2杂质:取20×20玻璃板两块,将番茄酱在玻璃板间涂布均观察杂质。
3.2可溶性固形物:按 GB/T 10786 规定的方法测定。
3.3 PH按 GB/T 10786 规定的方法测定。
3.4 番茄红素含量测定按 GB/T 14215 规定的方法测定。
3.5 色差值按 SN/T 1036 规定的方法测定4.检验规则4.1组批批的形成以同时进厂的产品为一批。
4.2取样10桶以下至少取1桶样,10桶以上开二桶,100桶以上按2%取样,开桶后用75%的酒精对包装物外部及取样工具消毒后,取样100g,再对外部消毒后扎口。
4.3判定规则4.3.1感观和可溶性固形物有一项不合格则整批不合格。
4.3.2供方必须随货提供该批产品的出厂检验报告单,有效的质量监督部门的定期监督检验报告单每年提供一次。
4.3.3本品所检项目全部符合要求方为合格。
5.包装、运输、贮存5.1包装产品单位包装应采用马口铁或无菌包装袋,查验产品生产日期、保质期符合规定,外包装应完好无损,无胀桶、漏料现象。
5.2运输使用清洁用具,防止污染,防止日晒雨淋。
5.3贮存产品应贮存在通风良好,避光之处。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
范围:本标准规定了番茄红素质量标准的检验方法和操作要求;本标准适用于番茄红素的质量检测。
引用标准:
GB/T 22249-2008 《保健食品中番茄红素的测定》
质量标准:
检验规程
1 番茄红素含量
1.1 原理
根据番茄红素易溶于二氯甲烷等溶剂的理化性质,试样经焦性没食子酸-二氯甲烷溶液提取,定容,过滤后进高效液相色谱仪,经反相色谱分离后,由紫外检测器检测,根据保留时间和峰面积进行定性和定量。
2.2 试剂与材料
2.1 乙腈:色谱纯。
2.2 甲醇:色谱纯。
2.3 二氯甲烷呢:分析纯。
2.4 焦性没食子酸:分析纯。
2.5 N,N-二甲基甲酰胺:分析纯。
2.6 番茄红素对照品:纯度≥95%,避光保存于-70℃冰箱。
2.7 焦性没食子酸-二氯甲烷溶液:称取6.0g焦性没食子酸,用二氯甲烷溶解并定容至100mL。
2.8 番茄红素对照品溶液:准确称量番茄红素0.001g(精确到0.0001g),置于10mL棕色容量瓶中,加焦性没食子酸-二氯甲烷溶液溶解并定容至刻度,混匀,此溶液临用现配。
由于番茄红素不稳定,使用前可先用液相色谱归一化法确定其纯度。
3. 仪器
3.1 高效液相色谱仪:附紫外检测器。
3.2 超声波清洗器。
4. 分析步骤
4.1 试样的制备
4.1.1 一般试样的制备
根据试样中番茄红素的含量,称取0.5g~2.0g均匀试样(精确称量至
超
声
提
取
3
m
i
n
后
,
加焦性没食子酸-二氯甲烷溶液定容至刻度,摇匀,过0.45μm滤膜。
滤液备
用。
4.1.2 微胶囊化试样的制备
根据试样中番茄红素的含量,称取0.5g~2.0g均匀试样置于25mL棕
色容量瓶中(精确称量至0.001g),加0.2g焦性没食子酸和5mL N,N-二甲
基甲酰胺后超声提取30min后,再用焦性没食子酸-二氯甲烷溶液定容至刻
度,摇匀,过0.45μm滤膜。
滤液备用。
4.2 标准曲线的制备
分别吸取番茄红素对照品溶液(2.8),用焦性没食子酸-二氯甲烷溶
液稀释并在棕色容量瓶中定容的浓度分别为0.2、0.5、1.0、5.0、10.0、
20.0μg/mL标准系列。
4.3 液相色谱参考条件
4.3.1 色谱柱:ODS C
18柱或ODS C
30
柱(适用于番茄红素几何异构体的分离
或含多种胡萝卜素试样的分析),150mm×4.6mm,5μm。
4.3.2 柱温:30℃。
4.3.3 紫外检测器:检测波长472nm。
4.3.4 流动相:甲醇+乙腈=50+50。
4.3.5 流速:1.0Ml/min。
4.3.6 进样量:10μL。
4.3.7 色谱分析:取标准溶液及试样溶液注入色谱中,以保留时间定性,
以试样峰面积或峰高与标准比较定量。
5 计算
试样中番茄红素的含量按式(1)进行计算:
c×V×1000
X=————————(1)
m×1000×1000
式中:
X——试样中的含量,单位为克每千克(g/kg);
c——根据标准曲线查得的番茄红素的浓度,单位为微克每毫升(μg/mL);
V——试样定容体积,单位为毫升(mL);
m——试样质量,单位为克(g)。
计算结果保留三位有效数字。
6 精密度
在重复性条件下获得的两次独立测定结果的绝对差值不得超过算术
宁波海逸生物科技有限公司
技术标准----原辅料质量标准及检验规程文件编号:STP-YL-017
番茄红素质量标准及检验规程版本/修改状态:A/0
复印份数:3份
编制人:毛雪雯审核人:李杰批准人:施建耀
编制日期:2011年 1月20日审核日期:2011年 1月20日批准日期:2011年 1月20日制定部门:技术管理部颁发部门:技术管理部执行日期:2011年 1月20日执行部门:技术管理部、质量管理部
在上述色谱条件下的色谱图见图1~图2。