药品生产对环境的要求(GMP培训教材)
gmp 附录一隔离器 背景环境 要求

gmp 附录一隔离器背景环境要求以GMP附录一隔离器背景环境为标题的文章随着医药行业的不断发展和进步,对于药品的生产质量和安全性要求也越来越高。
药品的生产过程中,存在着可能会受到外界环境污染的风险,因此需要在生产过程中采取一系列的措施来确保药品的质量和安全性。
GMP(Good Manufacturing Practice,良好生产规范)作为一种国际上公认的药品生产质量管理体系,规定了药品生产过程中的各项要求和标准。
GMP附录一隔离器就是其中之一。
GMP附录一隔离器是一种用于生产过程中隔离有害物质和保护操作人员安全的设备。
隔离器的主要作用是将操作人员与药品或其他有害物质隔离开来,防止操作人员接触到有害物质,同时也防止有害物质对外界环境的污染。
在药品生产过程中,一些药物可能具有较高的毒性或易感染性,如果操作人员接触到这些药物,可能会对其健康产生严重的危害。
因此,必须采取一些措施来保护操作人员的安全。
GMP附录一隔离器就是一种非常有效的措施之一。
GMP附录一隔离器主要由两部分组成:一个是隔离箱体,另一个是操作手套箱。
隔离箱体是一个密封的空间,用于将操作人员与药品隔离开来。
操作手套箱是一个装有手套的密封箱体,操作人员可以通过手套进行操作,而不会直接接触到药品。
隔离器的工作原理是通过负压控制来实现的。
在隔离箱体内部,通过空气流动的方式,将箱体内部保持在一个较低的压力状态,这样就可以防止有害物质逸出到箱体外部。
同时,通过过滤器对进入隔离器的空气进行过滤,确保空气中不含有有害物质。
在使用GMP附录一隔离器的过程中,需要严格遵守操作规程和操作要求。
操作人员必须进行专门的培训,熟悉隔离器的使用方法和操作流程。
在操作过程中,需要佩戴相应的个人防护装备,如手套、口罩和防护服等。
同时,需要对隔离器进行定期的维护和清洁,确保其正常工作和安全使用。
GMP附录一隔离器的使用不仅能够保护操作人员的安全,还能够保证药品的质量和安全性。
GMP与微生物--GMP培训资料

GMP与微生物GMP与微生物* 推行GMP目的:消灭污染、混药、差错* 污染的定义:当一个产品存在有不需要的物质时,它即受到污染。
* 污染的形式:尘粒污染、微生物污染尘粒污染* 尘粒污染:是指产品因混入不属于它的那些尘粒而变得不纯净。
尘埃、污物、棉绒、纤维以及我们的头发都是尘粒污染的潜在原因。
* 临床资料表明:如药品被7-2μm的尘粒污染了,尤其是静脉注射用药,可以导致热原反应、肺动脉炎、微血栓或异物肉芽肿等,严重的会致人死命。
微生物污染* 微生物污染是指因微生物产生、附着而给特定的环境带来的不良影响。
* 如果细菌得到了必要的养料、一定量的水份和合适的温度,它们就能迅速繁殖,它们繁殖的速度快得惊人。
通常一个细菌在仅仅24小时后可产生出281兆(百万)个的细菌。
传播污染的四大媒介* 空气* 水* 表面* 人不同环境中微生物量传播污染的四大媒介-空气* 空气:空气并不是产生污染的介质,但它是污染的危险携带者。
充满微生物的尘埃和水滴就是通过空气作为侵袭我们产品的媒介而成为重要的污染源的。
* 当我们谈话、咳嗽和打喷嚏时,被污了的水滴正是不断地从我们的呼吸道中放入我们的工作场所。
尘埃微粒正是通过空气流不间断地在我们整个药厂范围循环运行着。
* 请记住:空气中存在的尘埃微粒是含有微生物(细菌等)的。
因此,空气是侵袭我们产品的污染物的主要媒介。
这就是为什么要尽量保持空气洁净是极为重要的。
传播污染的四大媒介-水* 水:从理论上来讲,微生物在纯水中是不能生长的。
但是,所有的各类水,不管怎样仔细地蒸溜或过滤,总会含有一定量的可溶性有机物和盐类。
正是这些可溶性的物质可被微生物利用作为它们生长的养料源泉。
传播污染的四大媒介-水* 在生产过程中,水是应用最广泛的原料之一。
这就是为什么我们药厂的生产不仅必须仔细地控制用于生产过程的水,而且要控制好如何正确地用水来清洗我们的设备。
传播污染的四大媒介-表面* 表面包括:天花板、墙壁、地面、设备、容器、工具或桌子;* 由于空气中的湿度,所有表面都包上一层含水的薄膜。
2024年GEP与GMP培训教程

GEP与GMP培训教程一、引言在当今社会,随着科技的发展和人们对生活品质的追求,制药行业得到了迅猛发展。
然而,制药行业的竞争日益激烈,如何在竞争中脱颖而出,成为制药企业关注的焦点。
本文将为大家介绍GEP 与GMP培训教程,帮助大家了解制药行业的基本要求和规范,提高企业的竞争力。
二、GEP培训教程1.GEP概述GEP(GoodEngineeringPractice)是指良好的工程实践,它是一套针对制药行业工程技术人员的规范,旨在确保制药工程项目的设计、施工、运行和维护等环节都能满足药品生产的要求。
GEP培训教程旨在帮助工程技术员掌握GEP的基本知识和操作技能,提高工程项目的质量和效率。
2.GEP培训教程内容(1)GEP基础知识:介绍GEP的概念、起源、发展历程以及在我国的应用现状,使学员对GEP有一个全面的了解。
(2)GEP法规要求:分析我国GEP相关法规、政策,让学员明确GEP在制药行业中的地位和作用。
(3)GEP实施策略:探讨如何将GEP应用于制药工程项目,包括项目策划、设计、施工、验收、运行和维护等方面。
(4)GEP案例分析:通过实际案例,分析GEP在制药工程项目中的应用效果,使学员更好地理解GEP的实际意义。
(5)GEP质量管理:介绍GEP质量管理体系,包括质量策划、质量控制、质量保证和质量改进等方面,提高学员的质量管理水平。
(6)GEP操作技能:培训学员掌握GEP相关操作技能,如设备选型、工艺设计、施工管理等,提高工程项目的实施能力。
3.GEP培训教程的实施(1)培训对象:制药企业工程技术员、项目经理、质量管理人员等。
(2)培训方式:采用线上培训、线下培训相结合的方式,确保学员能够充分掌握GEP知识。
(3)培训时间:根据企业需求,制定合理的培训计划,确保培训效果。
(4)培训效果评估:通过考试、实操演练等方式,评估学员对GEP知识的掌握程度,确保培训质量。
三、GMP培训教程1.GMP概述GMP(GoodManufacturingPractice)是指良好的生产实践,它是一套针对制药行业的生产质量管理规范,旨在确保药品生产过程的质量和安全性。
2010版GMP附录1无菌药品培训教材

2010版GMP无菌药品附录
15章、81条、10471个字
本附录包含的各章节
第一章 范围 第二章 原则 第三章 洁净度级别及 监测 第四章 隔离操作技术 第五章 吹灌封技术 第六章 人员 第七章 厂房 第八章 设备 第九章 第十章 第十一章 第十二章 第十三章 第十四章 第十五章 消毒 生产管理 灭菌工艺 灭菌方法 无菌药品的最 终处理 质量控制 术语
第二章
原则
第六条 物料准备、产品配制和灌装或分装等操作 必须在洁净区内分区域(室)进行。 第七条 应当根据产品特性、工艺和设备等因素, 确定无菌药品生产用洁净区的级别。每一步生产操 作的环境都应当达到适当的动态洁净度标准,尽可 能降低产品或所处理的物料被微粒或微生物污染的 风险。 确定无菌药品生产用洁净区的级别应根据风险评 估结果,不应机械、教条地执行规定 强调无菌药品生产的环境应达到动态洁净度标准
第五章 吹灌封技术
第十七条 用于生产非最终灭菌产品的吹灌封设备 自身应装有A级空气风淋装置,人员着装应当符合 A/B级洁净区的式样,该设备至少应当安装在C级 洁净区环境中。在静态条件下,此环境的悬浮粒子 和微生物均应当达到标准,在动态条件下,此环境 的微生物应当达到标准。 用于生产最终灭菌产品的吹灌封设备至少应当安装 在D级洁净区环境中。 吹灌封技术是另一种先进的密闭生产技术,是鼓 励采用的生产方法
表面微生物 洁净度 级别 浮游菌 cfu/m3 沉降菌 (90mm) cfu /4小时 1 5 50 接触 (55mm) cfu /碟 1 5 25 5指手套 cfu /手套 1 5 -
A级 B级 C级
1 10 100
D级
200
100
50
-
第三章 洁净度级别及监测
《食品公司GMP培训》课件

随着食品产业的快速发展,如何确保生产过程中的卫生、安全和质量控制成为GMP面 临的重要挑战。
机遇
随着消费者对食品安全需求的增加,GMP的严格执行将为企业赢得市场信任,提升品 牌形象。
GMP发展建议
加强国际合作
积极参与国际食品安全标 准的制定和推广,提高我 国食品产业的国际竞争力 。
强化技术创新
审核标准与流程
现场审核
审核机构对企业的生产现场进行实地 审查,验证实际操作与文件的一致性 。
审核结论
审核机构根据审查情况出具审核结论 ,包括是否通过、整改要求等。
持续改进与优化
持续改进
对检查和审核中发现问题进行整改,不断优化生 产流程和操作规范。
定期组织内部自查,及时发现并纠正不符合GMP 要求的行为。
食品公司应建立严格的标识制度,确保每种食品都有明确的标识,包括名称、生 产日期、保质期等信息。此外,还应采取措施防止不同种类或不同批次的食品混 淆,如使用不同的存储容器、标签等。
保证产品质量
总结词
确保生产出的食品符合预定的质量标准。
详细描述
食品公司应建立完善的质量管理体系,确保生产出的食品符合预定的质量标准。这包括对原材料的质量控制、生 产过程的监控、产品的检验和储存等方面的要求。同时,还应及时处理不合格产品,防止其流入市场。
《食品公司gmp培 训》ppt课件
目录
CONTENTS
• GMP简介 • GMP基本原则 • GMP实施要求 • GMP检查与审核 • GMP案例分析 • GMP未来展望
01 GMP简介
GMP的定义
GMP是英文Good Manufacturing Practice的缩写,中文的意思是“良 好作业规范”,或是“优良制造标准 ”,是一种特别注重在生产过程中实 施对产品质量与卫生安全的自主性管 理制度。
GMP知识培训教材1

GMP知识培训教材1第一部分我们的使命一、健康、疾病与药品健康(health)的定义:人体各器官系统发育良好、功能正常、体质健壮、精力充沛,并具有健全的身心和社会习惯能力的状态。
通常用人体测量、体格检查和各种心理和生理指标来衡量。
疾病(disease)的定义:指人体在一定条件下,由致病因素所引起的一种复杂而有一定表现形式的病理过程。
药品的定义:药品是指用于预防、治疗、诊断人的疾病,有目的地调剂人的生理机能并规定有习惯症、用法和用量的物质,包括中药材、中药饮片、中成药、化学原料药及其制剂、抗生素、生化药品、放射性药品、血清疫苗、血液制品和论断药品等。
认识药品药品的特性:1.种类复杂性药品种类有十多种,具体品种,全世界大约有20000余种,我们目前中药制剂约5000多种,西药制剂约4000多种,由此可见,药品的种类复杂、品种繁多。
2.药品的医用专属性药品不是一种独立的商品,它与医学紧密结合,相辅相成。
患者只有通过大夫的检查诊断,并在大夫的指导下合理用药,才能达到防止疾病、爱护健康的目的。
3.药品质量的严格性药品直截了当关系到人们的躯体健康甚至生命存亡,因此,其质量不得有半点马虎。
我们必须确保药品的安全、有效、均一、稳固。
(1)安全性指按规定的习惯症和用法、用量使用药品后,人体产生毒副反应的程度。
(2)有效性指在规定的习惯症、用法和用量的条件下能满足预防、治疗、诊断人的疾病,有目的地调剂人的生理机能的要求。
(3)均一性指药品质量的一致性,要紧表现为物理分布方面的特性,是体现药品质量标准的质量特性。
(4)稳固性指药品质量的稳固程度,在规定的条件下保持其有效性和安全性的能力。
稳固性是药品的重要质量特点。
稳固性好,有效期就长,服用也方便。
药品生产的一样过程以及阻碍因素,见图1—1。
图1—1 药品生产的一样过程第三章我们的使命药品生产企业要紧由三部分组成:生产系统、质量系统和客户服务系统。
我们的使命确实是保证药品安全、有效、均一、稳固。
GMP一般生产区环境卫生管理规程
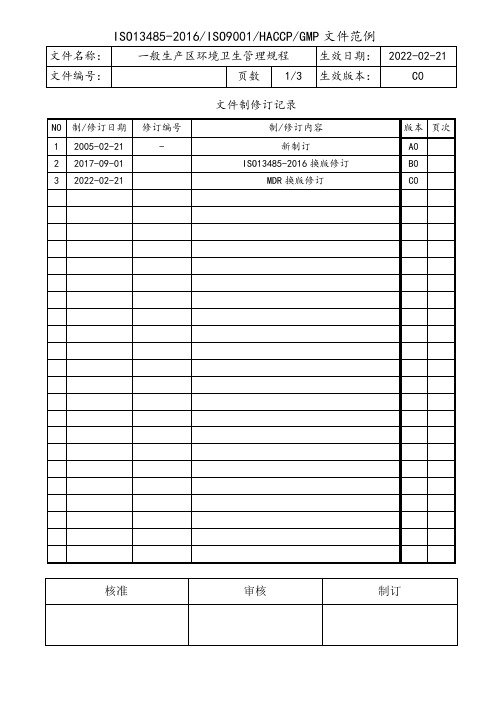
文件制修订记录1.0目的建立一般生产区的环境卫生管理规程,确保一般生产区的生产操作不受污染。
2.0范围一般生产区,提取和制剂车间的非洁净区。
3.0责任技术开发部、生产部、质量管理部、生产车间。
4.0管理内容4.1一般生产区环境卫生管理,主要包括厂房卫生管理,即地面、墙棚、灯具、门窗、操作台等的卫生管理。
4.2管理标准4.2.1厂房内壁、顶棚平整、光滑、清洁;门窗洁净,无浮尘,无霉斑,无渗漏,无不清洁的死角。
4.2.2地面光滑、平整、清洁、无积水,无杂物。
4.2.3厂房严密,无啮齿类动物及其它害虫。
设置电子捕虫装置,防止蚊蝇进入。
4.2.4原辅料、中间产品分类、定量、定点码放整齐,有明显的状态标记,可以有效地防止交叉污染和差错。
4.2.5一切非生产用品不得带入生产厂房,不得在生产厂房内吸烟、吃饭、睡觉、会客及大声喧哗,不得从事与生产无关的活动。
4.2.6楼道、走廊、电梯间不得放置任何物品,不得堆放成品及中间产品,保持运输通道的清洁、畅通。
4.2.7人流、物流能严格分开,有明显的标记,人员、物料能在规定的通道出入,没有穿行操作室。
4.2.8生产中的废弃物应装在密闭的容器内存放,每日能及时清理到规定的废弃物堆放站。
4.2.9生产区内设置相应的清洁间,卫生工具齐全,消毒措施完备,通风良好,工房清洁。
用具使用后及时处理干净。
清洁工具及清洁剂(消毒剂)要妥善存放,不能造成对药品生产环境的污染。
4.3管理要求4.3.1根据本公司“卫生管理规程”和本区域的卫生管理标准,由技术开发部负责制定“一般生产区厂房清洁规程”、“卫生间清洁规程”等清洁规程。
4.3.2生产厂房应时刻保持清洁,各项清洁规程,按GMP要求,内容包括:清洁方法、程序,间隔时间,使用的清洁剂或消毒剂、清洁工具的清洁方法和存放地点,同时分别制定出每日、每周和每月的清洁内容。
4.3.3厂房按规定要求进行清洁后,及时做好清洁记录。
4.3.4生产厂房的清洁卫生管理由生产车间主任负责,并指定车间卫生清洁员专人负责更衣室、办公室、参观走廊及其公用场所的清洁工作;生产作业区由所在操作室的操作人员承担清洁工作,按规定要求进行清洁,并能符合标准。
GMP与微生物--GMP培训资料

GMP与微生物GMP与微生物* 推行GMP目的:消灭污染、混药、差错* 污染的定义:当一个产品存在有不需要的物质时,它即受到污染。
* 污染的形式:尘粒污染、微生物污染尘粒污染* 尘粒污染:是指产品因混入不属于它的那些尘粒而变得不纯净。
尘埃、污物、棉绒、纤维以及我们的头发都是尘粒污染的潜在原因。
* 临床资料表明:如药品被7-2μm的尘粒污染了,尤其是静脉注射用药,可以导致热原反应、肺动脉炎、微血栓或异物肉芽肿等,严重的会致人死命。
微生物污染* 微生物污染是指因微生物产生、附着而给特定的环境带来的不良影响。
* 如果细菌得到了必要的养料、一定量的水份和合适的温度,它们就能迅速繁殖,它们繁殖的速度快得惊人。
通常一个细菌在仅仅24小时后可产生出281兆(百万)个的细菌。
传播污染的四大媒介* 空气* 水* 表面* 人不同环境中微生物量传播污染的四大媒介-空气* 空气:空气并不是产生污染的介质,但它是污染的危险携带者。
充满微生物的尘埃和水滴就是通过空气作为侵袭我们产品的媒介而成为重要的污染源的。
* 当我们谈话、咳嗽和打喷嚏时,被污了的水滴正是不断地从我们的呼吸道中放入我们的工作场所。
尘埃微粒正是通过空气流不间断地在我们整个药厂范围循环运行着。
* 请记住:空气中存在的尘埃微粒是含有微生物(细菌等)的。
因此,空气是侵袭我们产品的污染物的主要媒介。
这就是为什么要尽量保持空气洁净是极为重要的。
传播污染的四大媒介-水* 水:从理论上来讲,微生物在纯水中是不能生长的。
但是,所有的各类水,不管怎样仔细地蒸溜或过滤,总会含有一定量的可溶性有机物和盐类。
正是这些可溶性的物质可被微生物利用作为它们生长的养料源泉。
传播污染的四大媒介-水* 在生产过程中,水是应用最广泛的原料之一。
这就是为什么我们药厂的生产不仅必须仔细地控制用于生产过程的水,而且要控制好如何正确地用水来清洗我们的设备。
传播污染的四大媒介-表面* 表面包括:天花板、墙壁、地面、设备、容器、工具或桌子;* 由于空气中的湿度,所有表面都包上一层含水的薄膜。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
药品生产对环境的要求上海仁虎制药股份有限公司陆进方2008年11月药品生产对环境的要求我们在以前的培训中已经讲过,药品是一类用于预防、诊断、治疗疾病并具有调节人体机能的特殊商品。
药品具有商品的一般属性,但更有其特殊性。
药品的特殊性主要表现在药品所具有的专属性、两重性、时效性和质量控制严格性等四个方面。
正因为如此,决定了我们必须对药品的生产环境提出特殊的要求,药品生产环境必须符合所生产药品的工艺和质量特性的要求。
今天,我们就来共同探讨一下,药品生产对其生产环境究竟有何特殊要求。
我们主要分为药品生产企业的厂址选择、药品生产环境的一般要求和各剂型产品对生产环境的特殊要求三个部分来讨论。
第一部分药品生产企业的厂址选择2001年我国发布了关于新版的《洁净厂房设计规范》(GB50073-2001)作为工业生产用洁净厂房设计的国家统一标准,标准对工业洁净厂房的环境选择进行了规定。
同时,《制药企业GMP实施与认证指南》中,对药品生产企业的厂址选择也作出了相应的规定。
下面我们来看一下,药品生产企业在选择厂址时应当考虑哪些因素?1、应在大气含尘量小、含菌浓度低、无有害气体、自然环境好、对药品质量无有害因素、卫生条件较好的区域。
表1 大气中含尘浓度表2 大气含尘浓度平均值(大于或等于0.5µm,pc/L)2、应远离铁路、码头、机场、交通要道以及散发大量粉尘和有害气体的工厂(如化工厂、染料厂及屠宰厂等)、贮仓、堆场等有严重空气污染、水质污染、振动和噪音干扰的区域。
如不能远离严重空气污染区,则应位于其最大频率风向上风侧,或全年最小频率风向下风侧。
表3 天津地区的大气含尘计重浓度3、排水良好,应无洪水淹没危险。
4、目前和可预见的市政区域规划,不会使厂址环境产生不利于药品质量的影响。
5、水、电、燃料、排污、物资供应和公用服务条件较好或所存在的问题在目前和今后发展时能有效、妥善地解决。
在根据上述原则选择厂址的同时,应当经过技术经济方案的比较论证后,才能确定最终厂址。
第二部分药品生产环境的一般要求一、药品生产企业的总平面布置:药品生产企业的总平面布置必须遵循以下原则:1.药品生产企业的总平面布置在遵循国家有关工业企业总体设计原则外,还应按照不对药品生产产生污染,营造整洁的生产环境的原则确定。
2.生产、行政、生活和辅助区的总体布局应合理、不得互相妨碍。
3.生产厂房应布置在厂区环境清洁区域,厂区的地面、路面及运输不应对药品的生产造成污染。
4.药品生产厂房与市政交通干道之间距离不宜小于50m。
5.对于兼有原料药和制剂的药品生产企业,应考虑产品的工艺特点和防止生产时发生交叉污染,合理布局、间距恰当。
原料药生产区应置于制剂生产区的下风侧,青霉素等高致敏性药品的生产厂房的设置应严格考虑与其他产品的交叉污染。
6.在符合消防安全和尽量减少互相交叉污染的原则下,宜减少独立厂房幢数,建立联合厂房,以减少厂区道路及其造成的污染,减少厂区运输量和缩短运输线路。
但生产青霉素类等高致敏性药品必须使用独立的厂房与设施;避孕药品的生产厂房应与其他药品生产厂房分开;生产用菌毒种和非生产用菌毒种、生产用细胞与非生产用细胞、强毒与弱毒、死毒与活毒、脱毒前与脱毒后的制品和活疫苗与灭活疫苗、人血液制品、预防制品等的加工或灌装不得同时在同一生产厂房内进行,其贮存要严格分开。
7.危险品库应设于厂区安全位置,并有防冻、隆温、消防措施;麻醉药品和剧毒药品应设专用仓库,并有防盗措施。
8.动物房的设置应符合国家颁布的有关规定,并有专用的排污和空调设施。
9.厂区布置和主要道路应贯彻人、物流分流的原则,尽量避免相互交叉。
厂区道路面应选用整体性好、灰尘少的材料,如沥青、混凝土。
厂房与道路之间应有一定距离的卫生缓冲带,缓冲带可种植草坪,严禁种花,树木周围以卵石覆盖土壤,绿化设计做到“土不见天”。
10.厂房周围宜设环形消防车道(可利用交通道路),如有困难时,可沿厂房的两个长边设置消防通道。
11.药品生产厂房周围不宜设置排水明沟。
12.车辆的停车场应远离药品生产厂房。
13. 生产废弃物的回收应独立设置。
二、药品生产区域的环境参数:(一)一般规定1、药品生产区域的环境参数主要包括空气洁净度(尘粒数和微生物数)、温度和湿度、新鲜空气量、压差、照度、噪声等。
2、为了保证药品生产质量、防止生产环境对药品的污染,生产区域必须满足规定的环境参数标准。
3、药品生产区域应以空气洁净度(尘粒数和微生物数)为主要控制对象,同时还应对其温度、湿度、新鲜空气量、压差、照度、噪声等参数作出必要的规定,其中至少应对温度、湿度、压差、悬浮粒子、微生物进行验证。
4、环境空气中不应有不愉快气味以及有碍药品质量和人体健康的气味。
(二)环境参数的设计标准1、药品生产洁净室(区)空气洁净度分为四个等级表4 洁净室(区)空气洁净度级别表药品生产环境对洁净度的具体分区要求如下:100级洁净区:最终灭菌药品如大输液的灌封工序;非最终灭菌药品的药液的配制(灌装前不需除菌滤过的),注射剂灌封、分装、压塞,内包装材料最终处理后的暴露环境;质量标准中列有无菌检查项目的原料药的生产;灌装前不经除菌滤过的生物制品的配制、合并、灌装、冻干、加塞、添加稳定剂、佐料和灭活剂等工序。
10,000级洁净区:最终灭菌的药品如小容量注射剂的灌封,注射剂的稀配、滤过,内包装材料的最终处理(精洗)等;非最终灭菌药品的药液配制(灌封前需除菌滤过的);供角膜创伤、手术用滴眼剂的配制、灌封;灌装前经除菌滤过的生物制品的配制、合并、精制,添加稳定剂、佐料、灭活剂,除菌过滤、超滤、灌封等;体外免疫诊断试剂阳性血清分装、抗原-抗体分装等。
100,000级洁净区:最终灭菌药品的药液浓配、稀配;非最终灭菌药品轧盖,内包装材料最后一次精洗的最低要求;非最终灭菌口服液、深部组织创伤外用药品、眼用药品、腔道用药(除直肠用药外)生产的暴露工序;原料血浆的合并、非低温提取、分装前的巴氏消毒、轧盖以及制品的最终容器的清洗;口服制剂生产的暴露工序;发酵培养密闭系统的环境(暴露工序需无菌操作);霉联免疫吸附试剂配液、分装、干燥、包装;体外免疫试剂生产的暴露工序;深部组织创伤、大面积体表创面用生物制品的配制、灌装。
300,000级洁净区:口服固体制剂,最终灭菌的口服液体制剂,表皮外用药品,直肠用药,放射免疫分析药盒等生产的暴露工序;原料药生产暴露工序的最低要求。
2、温度和相对湿度:洁净室(区)的温度和相对湿度应与药品生产工艺相适应。
无特殊要求时,温度应控制在18~26℃,相对温度应控制在45%~65%。
3、压差:(1)洁净室必须维持一定的正压,可通过使送风量大于排风量的办法达到,并应有指示压差的装置。
(2)空气洁净度等级不同的相邻房间之间的静压差应≥5Pa,洁净室(区)与室外大气的静压差应≥10Pa,并应有指示压差的装置。
(3)工艺过程产生大量粉尘、有害物质、易燃易爆物质及生产青霉素类强致敏性药物,某些甾体药物,任何认为有致病作用的微生物的生产工序,其操作室与其相邻房间或区域应保持相对负压。
4、新鲜空气量:洁净室内应保持一定的新鲜空气量,其数值应取下列风量中的最大值:(1)非单向流洁净室应为总送风量的10%~30%,单向流洁净室应为总送风量的2%~4%;(2)补偿室内排风和保持正压值所需的新鲜空气量;(3)保证室内每人每小时的新鲜空气量不小于40m3。
5、照度:洁净室(区)应根据生产要求提供足够的照明。
主要工作室的照度宜为300Lux;在辅助工作室、走廊、气闸室、人员净化和物料净化用室可低于300Lux,但不宜低于150Lux;对照度不特殊要求的生产部位可设置局部照明,厂房应有应急照明设施。
6、噪声:洁净室内噪声级就符合下列要求:①动态测试时,洁净室的噪声级不宜大于75dBA;②静态测试时,乱流洁净室的噪声级不宜大于60dBA;层流洁净室的噪声级不宜大于65dBA。
洁净厂房的噪声控制设计必须考虑生产环境的空气洁净度要求,不得因控制噪声而影响洁净室的净化条件。
三、工艺布局的基本要求:1.工艺布局应按产品或剂型的生产工艺流程要求做到布置合理、紧凑,有利于生产操作,并能保证对生产过程进行有效的管理。
2.工艺布局要避免人、物流之间的混杂和交叉,防止引起污染和交叉污染,并符合下列要求:表5 洁净室内尘埃粒子来源分析表6 不同衣着、不同动作时的人体产尘(1)应分别设置人员和物料进、出生产区域的通道,必要时应设置极易造成污染的物料和废弃物的专用出口;(2)进入洁净区的人员必须有相应的净化用室和设施,其要求应与生产区洁净级别要求相适应;(3)进入洁净区的物料必须有与生产区洁净级别相适应的净化用室和设施,根据实际情况可采用物料清洁室、货淋(气闸室)或传递窗(柜)进入洁净区,进入非最终灭菌无菌药品生产区的原辅料、包装材料和其他物品必要时还应设置灭菌室或灭菌设施,但不得对洁净环境产生不良影响;(4)洁净区内物料传递输送路线尽量要短,减少折返;(5)生产中的废弃物不宜与物料进口合用一个气闸或传递窗(柜);(6)洁净区内的半成品不宜直接进入一般生产区,可采用传递窗(柜)、气闸或设置相应的设施进入一般生产区,传输带不得穿越不同洁净级别区域。
3.生产操作区内应设置必要的工艺设备和设施。
用于生产、贮存的区域不得作为非本区域内工作人员的通道。
4.人员和物料使用的电梯宜分开。
电梯不宜设施在洁净区内,必须设置时,电梯前设气闸室或采取确保洁净区空气洁净度的其他措施。
5.在满足工艺条件的前提下,为了提高净化效果,节约能源,有空气洁净度要求的房间尽量做到以下要求:(1)空气洁净度相同的房间或区域相对集中;(2)空气洁净度高的房间面积合理布置;(3)不同空气洁净度房间之间相互联系应有防止污染措施,如气闸室或传递窗(柜)等。
6.在药品洁净生产区域内应设置与生产规模相适应的备料室、原辅材料、中间体、半成品、成品存放区域。
存放区域内应安排待验区、合格品区和不合格品区,并按下列要求布置;(1)备料室、原辅材料存放区、中间体存放区、半成品存放区其空气洁净度与生产区空气洁净度相同;(2)备料室可视生产规模设置在仓库或生产车间内,并配备相应的称量室(区);(3)不合格中间体、半成品需设置专用回收间,其空气洁净等级宜同生产区的等级;(4)原辅材料、中间体、半成品存放区尽可能靠近与其相联系的生产区域,减少运输过程中的混杂和污染;(5)成品待验区与成品仓库区应有明显区别标志,不得发生混杂。
成品待验区可布置在生产区或入库前区。
四、洁净厂房的基本要求:1.建筑平面和空间布局应具有适当的灵活性。
洁净区的主体结构不宜采用内墙承重。
2.洁净厂房主体结构的耐久性应与室内装备、装修水平协调,并应具有防火、控制温度变形和不均匀沉陷性能。