G protein coupled receptors-systems (GPCRs).Illustrative advances from cannabinoid, chemokine and
细胞信号转导通路

Chromatin/Epigenetics Resources
Overview of Chromatin / Epigenetics
Chromatin regulation refers to the events affecting chromatin structure and therefore, transcriptional control of gene expression patterns. Epigenetics, specifically, refers to the heritable modifications which result in altered gene expression and are not known to be encoded in DNA. The nucleosome, made up of four histone proteins (H2A, H2B, H3, and H4), is the primary building block of chromatin. Originally thought to function as a static scaffold for DNA packaging, histones have more recently been shown to be dynamic proteins, undergoing multiple types of post-translational modifications (PTMs) and interacting with regulatory proteins to control gene expression. Protein acetylation plays a crucial role in regulating chromatin structure and transcriptional activity. Histone hyperacetylation by histone acetyltransferases (HATs) is associated with transcriptional activation, whereas histone deacetylation by histone deacetylases (HDACs) is associated with transcriptional repression. Hyperacetylation can directly affect chromatin structure by neutralizing the positive charge on histone tails and disrupting nucleosome-nucleosome and nucleosomeDNA interactions. In addition, acetylation creates binding sites for bromodomain-containing chromatin regulatory proteins (histone modification readers). Unlike acetylation, methylation does not alter the charge of arginine and lysine residues and is unlikely to directly modulate nucleosomal interactions required for chromatin folding. Methylated arginine and lysine residues are major determinants for formation of active and inactive regions of the genome. Methylation facilitates binding of chromatin regulatory proteins/histone modification readers that contain various methyl-lysine or methyl-arginine binding domains (PHD, chromo, WD40, Tudor, MBT, Ankyrin repeats, PWWP domains). Recruitment of co-activator and co-repressor proteins is dependent on the specific lysine residue that is modified. The modulation of chromatin structure is an essential component in the regulation of transcriptional activation and repression. One strategy by which chromatin structure can be modulated is through disruption of histone-DNA contacts by ATP-dependent chromatin remodelers, such as the NuRD, Polycomb, and SWI/SNF complexes, which have been shown to regulate gene activation/repression, cell growth, the cell cycle, and differentiation. Chromatin structure is also modulated through other PTMs such as phosphorylation of histone proteins, which affects association with DNA-interacting proteins and has been recently identified to play a role in coordinating other histone modifications. Furthermore, methylation of DNA at cytosine residues in mammalian cells affects chromatin folding and is a heritable, epigenetic modification that is critical for proper regulation of gene silencing, genomic imprinting, and development. Three families of mammalian DNA methyl-transferases have been identified, DNMT1/2/3, that play distinct roles in embryonic stem cells and adult somatic cells. In addition to the core histone proteins, a number of histone variants exist that confer different structural properties to nucleosomes and play a number of specific functions such as DNA repair, proper kinetochore assembly and chromosome segregation during mitosis, and regulation of transcription. Chromatin and epigenetic regulation is crucial for proper programming of the genome during development and under stress conditions, as the misregulation of gene expression can lead to diseased states such as cancer.
真菌胞内蛋白质提取方法的建立

[文章编号] 1671-587Ⅹ(2012)03-0590-05[收稿日期] 2011-08-15[基金项目] 国家自然科学基金资助课题(30910103903,30770120)[作者简介] 王 爽(1976-),女,吉林省长春市人,主治医师,医学博士,主要从事真菌分子学鉴定及耐药机制的研究。
[通信作者] 王 丽(Tel:0431-85619486,E-mail:wli99@jlu.edu.cn)真菌胞内蛋白质提取方法的建立王 爽1,2,姜兰香1,张 宇2,3,贺 丹2,田 庄2,高 嵩2,横山耕治4,王 丽2(1.吉林大学第二医院皮肤科,吉林长春130041;2.吉林大学白求恩医学院病原生物学系,吉林长春130021;3.吉林大学第二医院检验科,吉林长春130041;4.日本千叶大学真菌研究中心,日本千叶260-8673)[摘 要] 目的:研究不同的提取方法对白假丝酵母和茄病镰刀菌胞内蛋白质浓度、种类及数量的影响,为建立一种稳定可靠的真菌胞内蛋白质提取方法提供依据。
方法:分别以白假丝酵母菌和茄病镰刀菌为研究对象,培养时间为36、48和72h,超声波工作时间为20s、3min、5min,10min,超声波功率为332.5和475.0W,通过蛋白质浓度检测和SDS-PAGE蛋白质电泳分析,比较不同条件下提取的蛋白质浓度、种类及数量,观察不同因素对蛋白质提取效果的影响。
结果:2株真菌均在培养36h时提取胞内蛋白质的浓度最大,白假丝酵母在功率332.5W、超声3min时得到胞内蛋白质的数量和种类最多,茄病镰刀菌在功率332.5W、超声10min时得到蛋白质的数量和种类最多。
结论:使用超声波法与玻璃珠研磨法结合提取胞内蛋白质,从菌株的培养时间、超声波工作时间和功率3方面建立了真菌胞内蛋白质的最佳提取方法。
[关键词] 白假丝酵母;茄病镰刀菌;超声波;培养时间[中图分类号] R379.2 [文献标志码] AEstablishment of intracellular protein extractionmethods from fungusWANG Shuang1,2,JING Lan-xiang1,ZHANG Yu2,3,HE Dan2,TIAN Zhuang2,GAO Song2,KOJI Yokoyama4,WANG Li 2(1.Department of Dermatology,Second Hospital,Jilin University,Changchun 130041,China;2.Department of Pathogen Biology,Norman Bethune College of Medicine,Jilin University,Changchun130021,China;3.Department of Laboratory,Second Hospital,Jilin University,Changchun130041,China;4.Medical Mycology Research Center,Chiber University,Chiba 260-8673,Japan)Abstract:Objective To study the effect of different cultivate time,ultrasonic time and ultrasonic power on theconcentration,type and quantity of the intracellular protein from Candida albicans and Fusarium solina and toprovide the theoretical basis for establishing a reliable and stable extraction method for intracellular protein fromfungus.Methods Candida albicans and Fusarium solina were cultured.The different parameters included theculture time(36,48,and 72h),ultrasonic time(20s,3min,5min,and 10min)and ultrasonic power(332.5Wand 475.0W).The concentrations,species and quantities of protein under different factors were detected by theprotein concentration detection and SDS-PAGE electrophoresis analysis.Results Both strains got the most quantityof protein when cultivated for 36h.The most quantity and species of Candida albicans were obtained when theultrasonic power was 332.5Wand ultrasonic time was 3min,but as to Fusarium solina,the correspondingparameters were 332.5Wand 10min,respectively.Conclusion The intracellular protein is extracted bycompositely using ultrasonic method and glass bead transformation method.The optimal extraction method of095第38卷 第3期2012年5月吉 林 大 学 学 报 (医 学 版)Journal of Jilin University(Medicine Edition)Vol.38No.3 May 2012intracellular protein from fungus is developed from the aspects of culture time,ultrasonic power and ultrasonicworking time of strains.Key words:Candida albicans;Fusarium solani;ultrasound;culture time 随着免疫力低下或免疫缺陷患者等易感人群的不断增加,条件致病性真菌感染的发生率呈上升趋势,严重威胁患者的健康和生命[1-2],真菌致病和耐药机制的研究已成为当前研究的热点问题。
藏红花素介导DKK3调控GSK-3β

◇基础研究◇摘要目的:探讨藏红花素(crocin )对阿尔兹海默症(Alzheimer's disease ,AD )小鼠认知能力的改善作用及机制。
方法:SD 大鼠海马区注射A β25-35建立AD 模型,随机分为AD 组、AD+L 、M 、H-crocin 组(10、20、40mg/kg )和AD+donepezil 组(1mg/kg 盐酸多奈哌齐),腹腔注射治疗4周,另设置Sham 组。
采用避暗实验、水迷宫实验评估大鼠学习、记忆能力,ELISA 测定大鼠血清A β含量,HE 染色和Tunel 染色确定大鼠海马区内病理改变及神经元细胞凋亡,免疫组化测定大鼠海马区Brdu 、Dcx 、NeuN 表达,Western blot 测定大鼠脑组织A β、DKK3、β-catenin 、p-GSK-3β/GSK-3β、Caspase-3、Bax 、Bcl-2蛋白表达。
结果:与Sham 组相比,AD 组大鼠的学习、记忆能力下降,血清A β含量升高,且海马区的病理改变严重,神经元细胞凋亡增加,Brdu 、Dcx 、NeuN 含量降低,A β、DKK3、p-GSK-3β/GSK-3β、Caspase-3、Bax 蛋白表达升高,β-catenin 、Bcl-2蛋白表达降低(P <0.01)。
与AD 组相比,给予不同剂量crocin 和donepezil 治疗后,AD 大鼠学习、记忆能力提高,血清A β含量降低,海马区的病理改变减轻,神经元细胞凋亡减少,Brdu 、Dcx 、NeuN 含量升高,A β、DKK3、p-GSK-3β/GSK -3β、Caspase-3、Bax 蛋白表达升高,β-catenin 、Bcl-2蛋白表达降低(P <0.05),crocin 的剂量依赖效应显著。
结论:crocin 通过减少神经元细胞凋亡,介导DKK3调控GSK-3β/β-catenin 通路来改善AD 大鼠认知损伤。
阿尔兹海默症发病相关蛋白互作网络构建 与通路分析

Hans Journal of Biomedicine 生物医学, 2018, 8(2), 16-26Published Online April 2018 in Hans. /journal/hjbmhttps:///10.12677/hjbm.2018.82003Protein-Protein-Interaction Networkand Pathway Analysis Relatedto Alzheimer’s DiseaseYuchen Xu1, Lin Wei2, Honglei Li31Department of Neurology, Xiangya Hospital, Central South University, Changsha Hunan2Xiangya School of Medicine, Central South University, Changsha Hunan3Department of Otorhinolaryngology, Third Xiangya Hospital, Central South University, Changsha HunanReceived: Mar. 21st, 2018; accepted: Apr. 2nd, 2018; published: Apr. 9th, 2018AbstractAlzheimer’s disease (AD) is one of the most common neurodegenerative disease, and its pathoge-nesis is not fully understood. This article derives transcriptome data of patients with AD from the Gene Expression database GEO (Gene Expression Omnibus), through which we obtain the AD-related risk gene set. Based on AD risk gene set, we construct a protein-protein-interaction network of AD. At the same time we used functional enrichment analysis and path analysis to ex-plore the biological processes and signal pathway involved in AD. Our work helps to elucidate the pathogenic mechanism of AD and provide guidance to the future prevention and treatment of AD.KeywordsAlzheimer’s Disease, Protein-Protein-Interaction Network, Functional Enrichment Analysis阿尔兹海默症发病相关蛋白互作网络构建与通路分析徐煜宸1,魏琳2,李泓磊31中南大学湘雅医院神经内科,湖南长沙2中南大学湘雅医学院,湖南长沙3中南大学湘雅三医院耳鼻喉科,湖南长沙收稿日期:2018年3月21日;录用日期:2018年4月2日;发布日期:2018年4月9日徐煜宸 等摘要阿兹海默症(Alzheimer’s diseas, AD)是最常见的神经退行性疾病,其发病机制尚未完全明确。
华中科技大学学术期刊分类目录(T-D)_最新权威版
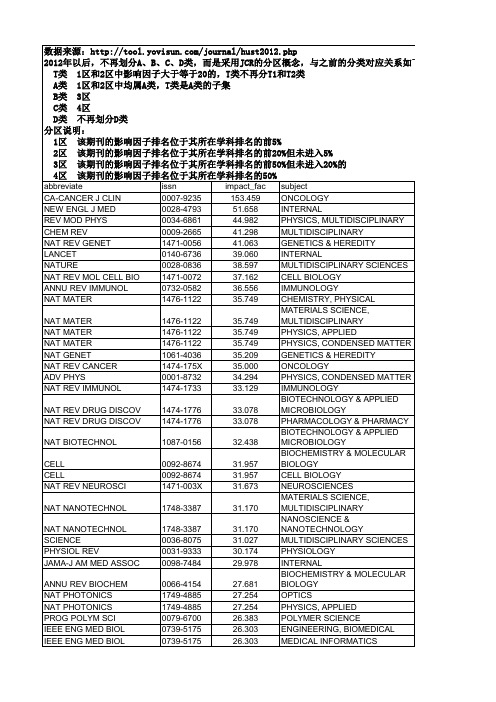
24.755 23.917 23.654 23.565 23.333 23.194 22.929 22.864 22.864 22.864 22.490 22.345 22.333 21.757 21.543 21.543 20.833 20.761 20.614 19.966 19.795 19.547 19.352 18.571 18.571 18.038 17.983 17.983 17.949 17.689 17.689 17.689 17.436 17.313 17.215 16.417 16.238 16.179 16.008 16.008 15.766 15.575 15.518 15.389 15.389 15.389 15.333 15.333 15.280 15.280 15.265 15.253 15.251 15.202
ONCOLOGY CLINICAL NEUROLOGY PLANT SCIENCES BIOCHEMICAL RESEARCH METHODS ASTRONOMY & ASTROPHYSICS MATERIALS SCIENCE, MULTIDISCIPLINARY PHYSICS, MULTIDISCIPLINARY BIOCHEMISTRY & MOLECULAR BIOLOGY CELL BIOLOGY MEDICINE, RESEARCH & EXPERIMENTAL MICROBIOLOGY PHARMACOLOGY & PHARMACY PHYSICS, PARTICLES & FIELDS CHEMISTRY, MULTIDISCIPLINARY PHARMACOLOGY & PHARMACY TOXICOLOGY CHEMISTRY, MULTIDISCIPLINARY CELL BIOLOGY NEUROSCIENCES INFECTIOUS DISEASES IMMUNOLOGY PHYSIOLOGY PHYSICS, MULTIDISCIPLINARY BEHAVIORAL SCIENCES NEUROSCIENCES ONCOLOGY CELL BIOLOGY DEVELOPMENTAL BIOLOGY ECOLOGY CHEMISTRY, MULTIDISCIPLINARY MAபைடு நூலகம்ERIALS SCIENCE, MULTIDISCIPLINARY NANOSCIENCE & NANOTECHNOLOGY GENETICS & HEREDITY MICROBIOLOGY MEDICINE, GENERAL & INTERNAL MICROBIOLOGY ASTRONOMY & ASTROPHYSICS MATERIALS SCIENCE, MULTIDISCIPLINARY BEHAVIORAL SCIENCES NEUROSCIENCES NEUROSCIENCES PSYCHOLOGY CLINICAL NEUROLOGY ECOLOGY EVOLUTIONARY BIOLOGY GENETICS & HEREDITY CHEMISTRY, PHYSICAL PHYSICS, CONDENSED MATTER BIOCHEMISTRY & MOLECULAR BIOLOGY CELL BIOLOGY PSYCHOLOGY MEDICINE, GENERAL & INTERNAL NEUROSCIENCES CARDIAC & CARDIOVASCULAR SYSTEMS
溶出度方法学验证-7

– Quats will ion pair with carboxylic acids at 2 pH’s units above pKa
13
Specific problems with SLS
• Use in combination only with Sodium Phosphate for 6.8 pH not Potassium Phosphate—precipitates at room temp
– Anionic
– Non-ionic
Evaluate different surfactants [quantitative]
4
Official Statements
FDA
• A dissolution medium containing surfactant can better simulate the environment of the gastrointestinal tract than a medium containing organic solvents or other nonphysiological substances, making the dissolution test conditions more useful in evaluating drug quality.
• Polysorbates (Tween™) • Sodium dodecyl sulfate (sodium lauryl sulfate) • Lauryl dimethyl amine oxide • Cetyltrimethylammonium bromide (CTAB) • Polyethoxylated alcohols • Polyoxyethylene sorbitan • Octoxynol (Triton X100™) • N, N–dimethyldodecylamine– N–oxide • Hexadecyltrimethylammonium bromide (HTAB)
PRRS
Porcine reproductive and respiratory syndrome virus as a vector:Immunogenicity of green fluorescent protein and porcine circovirus type 2capsid expressed from dedicated subgenomic RNAsYanlong Pei a ,Douglas C.Hodgins a ,Jiaqiang Wu a ,1,Siao-Kun W.Welch b ,Jay G.Calvert b ,Gang Li c ,Yijun Du d ,Cheng Song a ,d ,Dongwan Yoo d ,⁎aDepartment of Pathobiology,University of Guelph,Guelph,Ontario,Canada N1G 2W1bP fizer Animal Health,Kalamazoo,MI 49001,USA cInstitute of Animal Health and Husbandry,Chinese Academy of Agricultural Sciences,Beijing,China dDepartment of Pathobiology,University of Illinois at Urbana-Champaign,2001South Lincoln Ave,Urbana,IL 61802,USAa b s t r a c ta r t i c l e i n f o Article history:Received 31January 2009Returned to author for revision 3March 2009Accepted 31March 2009Available online 6May 2009Keywords:PRRSVReverse geneticsForeign gene expression vector Vaccine vector Nidovirus ArterivirusPorcine reproductive and respiratory syndrome virus (PRRSV)is the causative agent of PRRS,which is characterized by late-term abortions in sows and respiratory disease in young ing an infectious cDNA clone of North American PRRSV strain P129,the viral genome was engineered to transcribe an additional subgenomic RNA initiating between non-structural and structural genes.Two unique restriction sites and a copy of the transcription regulatory sequence for ORF6(TRS6)were inserted between ORFs 1b and 2a,yielding a general purpose expression vector.The enhanced green fluorescent protein (GFP)gene was cloned between the unique sites such that the inserted gene was transcribed from TRS2which was located upstream within ORF1b,while the copy of TRS6drives ORF2a/b transcription.Upon transfection of cells with this plasmid,PRRSV infection was initiated and progeny virus “P129-GFP ”was obtained.Cells infected with P129-GFP showed fluorescence and the inserted gene was phenotypically stable for at least 37serial in vitro passages.Subsequently,a capsid (C)protein gene was cloned from porcine circovirus type 2(PCV2)recovered from an outbreak of porcine multisystemic wasting syndrome (PMWS)and inserted into the PRRSV infectious clone vector,generating virus “P129-PCV ”.To determine the immunogenicity of the recombinant viruses,pigs were immunized intramuscularly with P129-WT (wild-type),P129-GFP,or P129-PCV2.By 5weeks post-infection,speci fic antibody responses to GFP and PCV2capsid were elicited.This is the first report of foreign gene expression using PRRSV from dedicated subgenomic RNAs and demonstrates the potential use of PRRSV as a vaccine vector for swine pathogens.©2009Elsevier Inc.All rights reserved.IntroductionPorcine reproductive and respiratory syndrome (PRRS)is an emerged and re-emerging disease in pigs.The disease was first recognized in Germany and the US almost simultaneously in the late 1980s and has since spread globally to most pork producing countries (Keffaber,1989;Ben field et al.,1992;Albina,1997).PRRS is characterized by abortions and mummi fied fetuses in sows,and respiratory distress with poor growth in young pigs.The disease is mild in gilts and boars,but the virus (PRRSV)persists in semen and thus can be transmitted widely by arti ficial insemination.Since its emergence,PRRS has become one of the most economically important diseases in the swine industry.Modi fied-live vaccines are available,but safety and limited ef ficacy areongoing concerns.No speci fic treatment is available for PRRS,and thus the economic losses are enormous.Numerous isolates of PRRSV representing many geographical regions have been sequenced,revea-ling the existence of two distinct genotypes of PRRSV:European (type I)and North American (type II).The two genotypes share overall sequence identity of 63%and differ antigenically as well as genetically (Nelson et al.,1993;Meng et al.,1995;Wootton et al.,2000).PRRS virus is an enveloped,single-stranded,positive-sense RNA virus belonging to the family Arteriviridae and along with the Coronaviridae family,forms the order Nidovirales .The PRRSV genome is approximately 15kb in size and includes the 5′cap structure and 3′polyadenylated tail (Sagripanti et al.,1986;Wootton et al.,2000).The genome consists of nine genes:open reading frames (ORFs)1a,1b,2a,2b,3,4,5,6,and 7.The 5′three-quarters of the genome consists of two slightly overlapping ORFs,1a and 1b,and they are translated directly from the genome-sense RNA.ORF1b is expressed as a fusion protein with ORF1a by a frame-shifting mechanism,and the ORF1a and ORFla/b proteins are auto-cleaved by viral proteases into at least 13cleavage products that are theVirology 389(2009)91–99⁎Corresponding author.E-mail address:dyoo@ (D.Yoo).1Current address:Shandong Key Laboratory of Animal Disease Control and Breeding,Shandong Academy of Agricultural Sciences,Jinan,Shandong,China.0042-6822/$–see front matter ©2009Elsevier Inc.All rights reserved.doi:10.1016/j.virol.2009.03.036Contents lists available at ScienceDirectVirologyj o u r n a l ho m e p a g e :w w w.e l s ev i e r.c o m /l o c a t e /y v i r onon-structural proteins Nsp1α,Nsp1β,and Nsp2through Nsp12.The Nsps are thought to be involved in genome replication and subgenome transcription(van Dinten et al.,1999;van Marle et al.,1999).The viral structural proteins are encoded by ORFs2a,2b,and3through7which are located downstream of ORFs1a and1b.These genes are expressed as a3′-coterminal nested set of subgenomic(sg)RNAs(de Vries et al., 1990).The5′untranslated leader sequences of the sgRNAs are derived from the5′end of the viral genome and fused to the body segments of the sgRNAs at conserved hexanucleotide motifs[5′UCAAC(U/C)3′] located immediate upstream of every transcription unit(de Vries et al., 1990;den Boon et al.,1996).The conserved hexanucleotide motif and poorly conservedflanking sequences form secondary structures in the sgRNAs that make up the transcriptional regulatory sequences(TRS)that are necessary for sgRNA formation(Pasternak et al.,2000).With the exception of sgRNA7,sgRNAs are structurally polycistronic but,with the exception of sgRNA2a/b,functionally monocistronic as only the5′most proximal gene of each sgRNA is translated.ORFs2a,2b,and3through7 code for GP2(glycoprotein2),E(envelope),GP3,GP4,GP5,M (membrane)and N(nucleocapsid)proteins,respectively,and they make up virion particles(Meulenberg et al.,1995).The recent development of infectious clones for PRRSV has allowed specific alterations of viral genomes and generation of mutant viruses(Yoo et al.,2004).However,manipulation of arterivirus genomes is complicated by the condensed organization of the viral genome.In the case of the European genotype of PRRSV,each gene overlaps slightly except for ORFs 1b and2a(Meulenberg et al.,1993),although the overlap of genes seems less important for virus replication and growth for equine arteritis virus (de Vries et al.,2000).For the North American genotype,structuralgenes Fig.1.Genomic organization of P129-WT,P129-GFP,and P129-PCV2.(A)Genomic organization and sequence of the region surrounding the ORF1b/ORF2junction of the unmodified “wild-type”P129strain of PRRSV(P129-WT).The boxed sequence indicates the core hexanucleotide of TRS2.Amino acids indicate translated sequences of polyprotein1a/b(ORF1b),the GP2protein(ORF2a),and the E protein(ORF2b).The initiation codons are indicated in bold,and the E protein sequence is italicized.Numbers in parenthesis indicate genomic sequence positions.Stars indicate translation stops.(B)P129-GFP.Unique AflII and Mlu I sites and a copy of TRS6were introduced into the non-coding region between ORF1b and ORF2a,creating expression vector pCMV-S-P129-1bMCS2.The GFP gene was amplified by PCR and inserted between the AflII and Mlu I restriction sites.(C)P129-PCV2.The PCV2 capsid gene was amplified by PCR and cloned between the AflII and Mlu I restriction sites.92Y.Pei et al./Virology389(2009)91–99also overlap with the exceptions of ORFs 1b and 2a,and ORFs 4and 5(Nelsen et al.,1999;den Boon et al.,1991;Snijder and Meulenberg,1998).Thus,the entire genome of the North American type PRRSV possesses only 4short non-coding regions:191nucleotides of 5′untranslated region (UTR),1nucleotide between ORF1b and ORF2a,10nucleotides between ORF4and ORF5,and 151nucleotides of 3′UTR upstream of the polyadenylation tail.The 5′and 3′UTRs contain genome replication and transcription signals,and therefore offer limited sites for gene insertion and manipulation.The presence of overlapping genes hampers mutational analysis of the N-and C-termini of the structural proteins and also makes it dif ficult to insert heterologous genes into the viral genome.In the present study,we used a genomic cDNA clone of PRRSV (Lee et al.,2005)to generate a vector for foreign gene expression from a dedicated subgenomic transcription unit inserted in the region between the structural and non-structural genes.The vector was used to express GFP in vitro and in vivo .The inserted gene was tolerated by the virus,stable for at least 37passages in cell culture,and induced antibodies to GFP in young pigs.Using this approach,we also generated a recombinant PRRSV expressing the capsid protein gene of porcine circovirus type 2(PCV2).PCV2is a small DNA virus in the Circoviridae family,with a genome ofonly 1.76kb.PCV2ORF1is essential for viral DNA replication (Mankertz et al.,1998;Fenaux et al.,2000),while ORF2encodes the capsid protein containing type-speci fic epitopes that are believed to be important for virus neutralization (Nawagitgul et al.,2000;Fenaux et al.,2004).Accumulating evidence suggests a major role for PCV2in postweaning multisystemic wasting syndrome (PMWS)and porcine dermatitis and nephropathy syndrome (PDNS)(Hasslung et al.,2005).These syndromes cause serious economic impacts in the swine industry today (Chae 2005).We showed the recombinant PRRSV P129-PCV2induced an anti-PCV2antibody response in immunized pigs in the presence of maternal antibodies to PCV2.Our vector construction may be applicable not only for PRRSV,but also for other members of the families Arteriviridae and Coronaviridae .ResultsDevelopment of PRRSV as an expression vector for GFPTo explore the possibility of developing PRRSV as a gene expression vector,the GFP gene was inserted between the stop codon ofORF1bFig.2.(A)Restriction patterns of P129-WT (lane 1),P129-GFP (lane 2),and P129-PCV2(lane 3)genomic clones generated by Sma I digestion.Fragments of 590bp,4736bp and 10,779bp are expected from all three clones.The fourth fragment varies in size depending on the gene inserted at the non-structural and structural gene junction.Relative to the P129-WT fragment (2787bp,lane 1),the GFP-containing fragment from P129-GFP is 766bp larger (3553bp,lane 2)and the PCV2capsid-containing fragment from P129-PCV2is 754bp larger (3541bp,lane 3).(B –E)Recovery of recombinant PRRSV foci from full-length genomic clones.(F)Integration of the GFP gene in P129-GFP viral genome.Genomic RNA was extracted from P129-GFP virus and digested with DNase I prior to reverse transcription.Without reverse transcription,no products were ampli fied from P129-GFP using ORF7-speci fic PCR primers P129-7F and P129-7R (lane 1)or primers P129-1bF and P129-2aR that span the insertion site (lane 2).With reverse transcription,primers P129-7F and P129-7R ampli fied the ORF 7fragment (534bp)from both P129-WT (lane 3)and P129-GFP (lane 4).Using P129-1bF and P129-2aR,a product of 766bp larger (lane 6)than the product from P129-WT (lane 5)was ampli fied from P129-GFP.(G)Integration of the PCV2C gene in PRRSV.Genomic RNA from P129-WT (lanes 9,10,12)and P129-PCV2(lanes 7,8,and 11)was ampli fied using primers P129-1bF and pared to P129-WT (lane 9)a product that is 754bp larger was ampli fied from P129-PCV2(lane 7).Using PCV2C gene speci fic primers PCV2-F and PCV2-R,the expected 497bp product was generated from P129-PCV2(lane 8)but not from P129-WT (lane 10).Using PRRSV ORF4-speci fic primers P129-4F and P129-4R,the expected 567bp product was ampli fied from both P129-PCV2and P129-WT (lanes 11and 12,respectively).93Y.Pei et al./Virology 389(2009)91–99and the start codon of ORF2a (Fig.1A).This non-coding region is extremely short,comprising only one adenosine nucleotide.The TRS associated with ORFs 2a and 2b (TRS2;TGAACC)is positioned 26nucleotides upstream from the start of ORF2a and is embedded in ORF1b.Upon insertion of GFP to the region,TRS2will drive transcription of the GFP gene instead of ORFs 2a and 2b.Thus,a synthetic TRS (TTAACC)with flanking sequences derived from TRS6was introduced 22nucleotides downstream of GFP and 17nucleotides upstream from the ORF2a start (Fig.1B).TRS6was chosen because RNA secondary structure suggested that it was shorter than other PRRSV TRS elements and because the distance between the copy of TRS6driving ORF2a/b and the authentic TRS6ensured that potential intramolecular homologous RNA recombination would result in a non-viable (ORF2-5deleted)virus.The PRRSV genomic clone containing GFP was designated P129-GFP,and the insertion was con firmed by restriction patterns (Fig.2A,lane 2)and sequencing.MARC-145cells were transfected with P129-GFP and the production of virus was monitored daily for development of cytopathic effect (CPE)(Figs.2B,C,D).CPE was observed 4days post-transfection and the development of CPE was one day slower than for P129-WT.After three consecutive passages,P129-GFP virus was re-examined for GFP sequence integration in the viral genome (Fig.2F).While ORF7ampli fication products were identical in size for P129-WT (lane 3)and P129-GFP (lane 4),1bF and 2aR primers produced a larger size product from P129-GFP (lane 6)than from P129-WT (lane 5).The larger product was the expected size for correct insertion of GFP gene intothe viral genome.Replication of P129-GFP was slightly slower than that of P129-WT at passages 1to 3.However,plaques were comparable in size and morphology for P129-WT and P129-GFP,and the titers at passage 3were 5×105plaque forming units (PFU)/ml and 1×105PFU/ml,respectively.Fluorescence was evident in P129-GFP-infected cells (Fig.3A),demonstrating the expression of GFP during infection.To examine the genetic stability of recombinant PRRSV,P129-GFP was passaged 37times in MARC-145cells and GFP expression was monitored by fluorescence microscopy.Individual plaques of 20formed by P129-GFP were randomly chosen and examined for fluorescence.The selected plaques were all positive for fluorescence (Table 1),and sequencing of the viral RNA con firmed stability of the insert (data not shown).This data showed the genetic stability of P129-GFP after serial passages in cell culture and the stable expression of GFP.It also demonstrates that the region between ORF1b and ORF2a is a suitable site for foreign gene insertion for PRRSV.This was the first demonstration of the use of a nidovirus as an expression vector wherein the foreign gene is inserted in the region between the non-structural and structural protein codingsequences.Fig.3.Expression of GFP or PCV2capsid protein by P129-GFP and P129-PCV2in MARC-145cells.(A)Live cells infected with P129-GFP passage 3;(B)live cells infected with P129-GFP passage 37;(C)uninfected cells;(D)P129-PCV2passage 3fixed and stained with PCV2-speci fic antibody conjugated with FITC at 48h post-infection;(E and F)P129-PCV2infected cells fixed and co-stained with PCV2-speci fic antibody conjugated with FITC (E)or GP4protein-speci fic monoclonal antibody 169(F).Table 1Stability of GFP expression in P129-GFP virus.Virus Titer (PFU/ml)GFP expressing plaques Passage 38.3×10e620positive/20plaques Passage 372.4×10e720positive/20plaques94Y.Pei et al./Virology 389(2009)91–99Construction of PRRSV expressing PCV2capsidPCV2is associated with porcine multisystemic wasting disease (PMWS,now termed PCVAD [PCV-associated disease])and porcine dermatitis and nephropathy syndrome (PDNS).PCV2is transmitted by the oro-nasal route and shed in the bronchial secretions and feces,thus the transmission route is similar to that of PRRSV.The capsid (C)protein is the major antigen able to elicit protective immunity against PCV2.Thus,using the PRRSV vector system described above,an additional recombinant PRRSV was constructed to carry the PCV2C gene.A 702bp C gene was cloned by PCR from a lung tissue positive for PCV2.Sequencing of the C gene showed 99–100%amino acid identity to published sequences available in the GenBank database.The P129-PCV2clone was constructed by inserting the C gene into PRRSV in the same way as the GFP gene insertion for P129-GFP (Fig.1C).The insertion of the C gene was con firmed by restriction digestion pattern (Fig.2A,lane 3)and sequencing.The P129-PCV2recombinant virus was recovered from MARC-145cells by transfection (Fig.2E),and the insertion of the C gene in the viral genome was con firmed by RT-PCR of the viral RNA (Fig.2G).The titer of passage 3virus was 2×105PFU/ml and the plaque morphology was indistinguishable from P129-WT.Infection of cells with P129-PCV2and staining with PCV2-speci fic antibody produced distinct fluorescence (Fig.3E),which shows the expression of the C protein by P129-PCV2.The capsid protein of PCV2is arginine-rich and normally shuttles into the nucleus during PCV2replication (Liu et al.,2001),and similarly,the PRRSV N has also been shown to localize in the nucleus and nucleolus (Lee et al.,2006;Pei et al.,2008).Thus,in cells infected with P129-PCV2,the synthesis of legitimate PCV2capsid should be evident by the translocation of capsid into the nucleus.Thus,the C protein expression by P129-PCV2was con firmed by co-staining of virus-infected cells with PRRSV N protein-speci fic antibody and PCV2-speci fic antiserum (Fig.4).While the PRRSV N protein was found in the both cytoplasm and the nucleolus as usual (panel A),the PCV2capsid protein was speci fically localized to the nucleus and nucleolus (panel C)in the same cell,clearly demonstrating the expression of PCV2capsid protein by the recombinant PRRSVP129-PCV2.Fig.4.Co-expression of the PRRSV N protein (green)and the PCV2capsid protein (red)during infection of P129-PCV2.MARC-145cells were infected with P129-PCV2and stained 24h post-infection with PRRSV N-speci fic MAb SDOW-17(A and B)or PCV2-speci fic pig serum (C and D).Arrows indicate the PRRSV N protein in the nucleolus (panel A)in addition to the cytoplasm and the PCV2capsid protein in the nucleus and nucleolus (panel C).Panel E shows the merge of A and C.Panel F shows the merge of B and D.Table 2PCR primers and their genomic Sequence (5′–3′)aGenomic position b PurposePCV2-F cacggatattgtagtcctggt 1093–1114PCV2PCR test PCV2-R ccgcaccttcggatatactgtc1565–1586PCV2PCR testPCV2-orf2F gatgcttaagatgacgtatccaaggtggcg 1715–1734PCV2ORF2ampli fication PCV2-orf2R gtacacgcgtcattaagggttaagtcccccc 1031–1050PCV2ORF2ampli ficationP129-F1F aacagaagagttgtcgggtccac11,699–11,721P12911,783–12,055,ampli fication P129-F1R gctttcacgcgtccccacttaagttcaattcaggcctaaagttggttca 12,031–12,055Introduction of A flII and Mlu IP129-F2F gcgacgcgt gttccgtggcaacccctttaaccagagtttcagcggaaga atgaaatggggtctatacaaagcctcttcgaca 12,056–12,089P12912,056–12,697,ampli fication and introduction of Mlu I and TRS6P129-F2R aacagaacggcacgatacaccacaaa 13,819–13,844P12912,056–12,679,ampli fication P129-7F tcatccgattgcggcaaatg 14,724–14,743P129ORF7ampli fication P129-7R agaatgccagcccatca 15,242–15,258P129ORF7ampli fication P129-4F gtttcacctagaatggctg 13,213–13,231ORF4ampli fication P129-4R ccccaacatacttgaacattc 13,750–13,770ORF4ampli ficationP129-1bF ggtgaggactgggaggattac 11,921–11,941ORF1b –ORF2a,region ampli fication P129-2aRcagtacgtagcattggaacc12,758–12,777ORF1b –ORF2a,region ampli ficationa Restriction sites are underlined.TRS6and surrounding sequences are indicated in bold.bGenomic positions for PCV2primers were based on GenBank accession AF027217.Genomic position for P129primers were based on GenBank accession AF494042.95Y.Pei et al./Virology 389(2009)91–99Infection of pigs and antibody responses to GFP and PCV2capsid protein To determine antibody responses to GFP and the PCV2capsid protein,pigs were immunized with the recombinant PRRS viruses.Fifteen PRRSV-free pigs at 4weeks of age were randomly allotted to 3groups of 5pigs each.Animals were immunized twice on days 0and 21by intramuscular injection of 5×105PFU per animal with either P129-WT,P129-GFP,or P129-PCV2.Following inoculation,the animalswere maintained for 5weeks for clinical observation and serum collection.Clinical signs of PRRS were minimal and comparable in all 3groups (data not shown).Mild clinical signs were not unexpected,since the infectious cDNA clone used in these studies was not attenuated.Tonsil samples were collected at necropsy on day 35and assessed by RT-PCR for the presence of PRRSV ORF7using primers P129-7F and P129-7R (Table 2)as well as the GFP and PCV2capsid inserts using primers P129-1bF and P129-2aR.All 15pigs were positive for ORF7,indicating infection and persistence of PRRSV in the tonsils.PCR products from the ORF1b/ORF2a junction were not detected in these tonsil samples,possibly due to the much lower molar ratio of ORF1b-containing RNA template relative to ORF7-containing RNA in infected cells.Alternatively,it is possible that the GFP and PCV2capsid genes might have been unstable in vivo and lost in the inoculated pigs over time.To examine antibody responses in these pigs,ELISAs were conducted for PRRSV,GFP,and PCV2C protein.All pigs produced good levels of antibodies to PRRSV (Fig.5A),and the antibody titers were comparable among groups.P129-GFP elicited speci fic antibodies to GFP,whereas pigs immunized with P129-PCV2or P129-WT were negative for GFP (Fig.5B).The GFP antibodies increased following first immunization,and the second immunization at day 21boosted the antibody response somewhat (Fig.5B).Similarly,antibodies for PCV2C protein were detected in pigs immunized with P129-PCV2(Fig.5C).In these pigs,however,PCV2antibodies were detected at day 0in all 3treatment groups and tended to wane over time.These antibodies likely represent maternal antibodies taken up in colostrum shortly after farrowing at the farm of origin and prior to experimental infection.However,an increase in anti-PCV2antibodies was observed at 28days in the P129-PCV2group only (Fig.5C)due to boosting effects from the second immunization at day 21.In contrast,anti-PCV2antibodies in the other two treatment groups waned gradually from day 0until at least until day 35.To further determine the speci fic antibody responses to GFP and the PCV2C protein,Western blots were conducted using sera from these pigs.Serum from the P129-GFP group was reactive with GFP (Fig.6A),consistent with the ELISA data (Fig.5B).Similarly,serum from the P129-PCV2group showed a strong reaction with C pro-tein (Fig.6B).Weaker reactions were observed using sera from the P129-WT and P129-GFP groups,consistent with the presence of maternalantibodies.Fig.5.ELISA showing induction of speci fic antibodies in sera from pigs inoculated with P129-GFP,P129-PCV2,or P129-WT viruses.(A)antibodies to PRRSV N protein;(B)antibodies to GFP;(C)antibodies to PCV2Cprotein.Fig. 6.Western blots showing induction of speci fic antibodies in sera from pigs inoculated with P129-GFP,P129-PCV2,or P129-WT at day 0or day 35post-inoculation.Blots contain GFP protein (A)or PCV2virions (B).Arrows indicate positions of GFP and PCV2capsid protein.C denotes positive control (anti-GFP monoclonal antibody).M indicates molecular weight markers.The virus used to infect pigs that contributed to the serum pools is indicated above each lane.96Y.Pei et al./Virology 389(2009)91–99DiscussionThe primary target cell of PRRSV is the alveolar macrophage,and pigs are the only animal species known to be susceptible to PRRSV infection.Therefore,development of PRRSV as a vaccine vector would be useful for the delivery of porcine pathogen genes to the respiratory tract of the pig.Arterivirus genomes are organized in a complex way. Most genes overlap one another in different reading frames,making it difficult to engineer the genome for foreign gene insertion.An early approach involved engineering the3′terminal region of the N gene (Groot Bramel-Verheije et al.,2000).Modification of the3′terminal sequence of N gene was possible and caused only minimal effects on virus replication and growth.However,no more than7amino acids were inserted.More recently,Nsp2,a large product of proteolytic cleavage of the ORF1a and ORF1a/b polyproteins,was found to be remarkably heterogeneous in sequence,with several hypervariable regions.Therefore,the GFP gene was inserted in-frame into or between the hypervariable regions to create nsp2-GFP fusion proteins (Fang et al.,2006;Kim et al.,2007).This approach was successful and allowed for the production of recombinant viruses.However,in all cases the inserted GFP gene was not phenotypically stable and lost greenfluorescence after several passages in cell culture(Fang et al., 2006;Kim et al.,2007;Han et al.,2007).In the present study,we inserted GFP and the PCV2capsid genes into the short region separating the non-structural protein genes from the structural protein genes.In contrast to the nsp2-GFP fusion proteins described above,we expressed GFP as a separate transcription unit resulting in an additional sgRNA.This approach has the advantage of eliminating the need to alter the coding sequence of a viral gene,and also minimizes effects on expression and post-translational modification of viral gene products.As a result,our recombinant virus was stable for at least37cell culture passages without loss of the gene or the greenfluorescent phenotype.PRRSV is known to induce immune suppressive effects in pigs (Charerntantanakul et al.,2006)and can persist up to6months in infected pigs(Wills et al.,1997).For these reasons,co-infection of PRRSV with other pathogen such as PCV2can result in much more severe clinical outcome than either agent alone(Harms et al.,2001; Kim et al.,2003).Therefore,a dual-purpose vaccine capable of protecting pigs against both PRRS and PCV2would be advantageous. Our study demonstrates the potential of PRRSV as a viral vaccine vector.Prior to the purchase of animals for infection studies using P129-PCV2,all pigs were screened for PCV2by PCR.PCR is the gold standard for detection of PCV2,whereas antibody screening is not routinely conducted in diagnostic laboratories due to cross-reactivity between PCV2and PCV1which is ubiquitous and widely distributed in thefield (Magar et al.,2000).Although all pigs entering the present study tested negative for the presence of PCV2by PCR,antibodies were detected in sera collected on day0.These antibodies were most likely of maternal origin,since they decreased in concentration over the duration of the experiment in pigs receiving P129-WT or P129-GFP. Serological studies show that maternal antibodies for PCV2decay during thefirst2months of life(Rodríguez et al.,2002;Larochelle et al.,2003).Western blots and ELISA gave comparable results in this regard.In the current study,antibodies to PCV2only increased after day21of the study and did so only in pigs receiving P129-PCV2, suggesting that the increases were most likely specific responses to P129-PCV2vaccination.At the termination of the study(day35post-infection),tonsil samples in all3groups were positive for the PRRSV N gene by RT-PCR.The presence of the inserted GFP and PCV2genes in these same samples could not be confirmed using primersflanking the region of the gene insertion.No products were amplified,even from pigs infected with the P129-WT virus.Failure to amplify the ORF1b/ORF2a junction is likely the result of template RNA concentra-tions below the level of detection of the PCR assay,and is consistent with the observation that the copy number of ORF1b(present only on genomic RNA)is much lower than the copy number of ORF7(present on all sgRNAs as well as genomic RNA).In conclusion,a PRRSV gene expression vector was generated, capable of expressing a foreign gene from an additional transcription unit located in the region between the non-structural and structural genes of the virus.The recombinant PRRSVs expressing the GFP or PCV2capsid genes were generated and shown to replicate well in cell culture.The addition of766nt(GFP)or754nt(PCV2capsid)of foreign genetic material,representing approximately5%of the PRRSV genome,was tolerated with no evidence of compensatory deletions or rearrangements elsewhere in the genome.Pigs inoculated with these recombinant PRRSVs produced foreign gene specific antibodies.Our study demonstrates the potential of PRRSV to function as a vector for development of multivalent vaccines against swine diseases.Materials and methodsCells and virusesMARC-145African green monkey kidney cells(Kim et al.,1993) were maintained as previously described(Lee et al.,2003).Dulac porcine kidney cells,kindly provided by L.Babiuk(Vaccine and Infectious Disease Organization,SK,Canada),were grown in Modified Eagle's Medium(MEM)supplemented with5%fetal bovine serum (FBS;Gibco BRL),penicillin(100U/ml),and streptomycin(50μg/ml). Cells were maintained at37°C with5%CO2.Stocks of recombinant viruses derived from infectious clones were prepared by passaging three times on MARC-145cells.Titers of PRRSV were determined by standard plaque assays on MARC-145cells using6-well plates(35mm diameter)in duplicate.Plaques were stained with0.01%neutral red. For isolation of PCV2,lung tissues were obtained from a PCR-positive pig(Ontario18099)submitted to Animal Health Laboratory of the University of Guelph(Guelph,ON,Canada).The tissues were homogenized in PBS and thefiltrate was used to infect Dulac cells. At3days post-infection,cells were stained using a porcine circovirus hyperimmune serum(VMRD,Pullman,WA,USA)to confirm infection. On day4post-infection,cells were harvested and freeze–thawed three times.Cell debris was removed by centrifugation at5000×g, and the supernatant was stored at−80°C until use.Construction of a PRRSV expression vector and recombinant PRRSVsFor construction of the PRRSV expression vector,the regions flanking the ORF1b/ORF2a junction were amplified by PCR using the shuttle plasmid p2-7D4(containing genomic positions11,504to 15,395)as template.Two DNA products corresponding to positions 11,783to12,055and12,056to12,697were amplified.The primer set P129F1-F(containing an Eco47III site)and P129F1-R(containing AflII and Mlu I sites)was used for the upstream product(Table2).The primers set P129F2-F(containing Mlu I site and TRS6)and P129F2-R (containing a Bsr GI site)was used to amplify the downstream product(Table2).The twoflanking products were digested with Eco 47III–Mlu I and Mlu I–Bsr GI,respectively,and included in a three-way ligation with Eco47III–Bsr GI-digested full-length genomic cDNA clone pCMV-S-P129(Lee et al.,2003).The resulting construct pCMV-S-P129-1bMCS2contained a complete PRRSV genome with unique AflII and Mlu I sites and a copy of TRS6inserted between ORF1b and ORF2a.Transfection of MARC-145cells with this construct produced viable virus that replicated normally(data not shown).For insertion of foreign genes,pCMV-S-P129-1bMCS2was digested with AflII–Mlu I and ligated to either the GFP gene or the PCV2capsid gene into which AflII and Mlu I sites were introduced during PCR (Fig.1).Recombinant genomic clones were screened by Sma I digestion(Fig.2),and selected clones were sequenced to confirm the presence of insertions.The PCV2capsid protein gene was cloned97Y.Pei et al./Virology389(2009)91–99。
GAPDH脱氢酶结构
Crystal Structure of Glyceraldehyde-3-Phosphate Dehydrogenase1from Methicillin-Resistant Staphylococcus aureus MRSA252ProvidesNovel Insights into Substrate Binding and Catalytic MechanismSomnath Mukherjee,Debajyoti Dutta,Baisakhee Sahaand Amit Kumar Das⁎Department of Biotechnology, Indian Institute of Technology, Kharagpur,Pin-721302, West Bengal,India Received30March2010; received in revised form1July2010;accepted2July2010 Available online8July2010The dreaded pathogen Staphylococcus aureus is one of the causes of morbidity and mortality worldwide.Glyceraldehyde-3-phosphate dehy-drogenase(GAPDH),one of the key glycolytic enzymes,is irreversibly oxidized under oxidative stress and is responsible for sustenance of the pathogen inside the host.With an aim to elucidate the catalytic mechanism and identification of intermediates involved,we describe in this study different crystal structures of GAPDH1from methicillin-resistant S.aureus MRSA252(SaGAPDH1)in apo and holo forms of wild type,thioacyl intermediate,and ternary complexes of active-site mutants with physio-logical substrate D-glyceraldehyde-3-phosphate(G3P)and coenzyme NAD+.A new phosphate recognition site,“new P i”site,similar to that observed in GAPDH from Thermotoga maritima,is reported here,which is 3.40Åaway from the“classical P i”site.Ternary complexes discussed are representatives of noncovalent Michaelis complexes in the ground state.D-G3P is bound to all the four subunits of C151S.NAD and C151G.NAD in more reactive hydrate(gem-di-ol)form.However,in C151S+H178N.NAD, the substrate is bound to two chains in aldehyde form and in gem-di-ol form to the other two.This work reports binding of D-G3P to the C151G mutant in an inverted manner for the very first time.The structure of the thiaocyl complex presented here is formed after the hydride transfer.The C3 phosphate of D-G3P is positioned at the“P s”site in the ternary complexes but at the“new P i”site in the thioacyl complex and C1–O1bond points opposite to His178disrupting the alignment between itself and NE2of His178.A new conformation(Conformation I)of the209–215loop has also been identified,where the interaction between phosphate ion at the“new P i”site and conserved Gly212is lost.Altogether,inferences drawn from the kinetic analyses and crystal structures suggest the“flip-flop”model proposed for the enzyme mechanism.©2010Elsevier Ltd.All rights reserved.Edited by G.Schulz Keywords:glyceraldehyde-3-phosphate dehydrogenase;ground-state Michaelis complex;thiaocyl intermediate;new P i site;“flip-flop”mechanismdoi:10.1016/j.jmb.2010.07.002J.Mol.Biol.(2010)401,949–968Available online at *Corresponding author.E-mail address:amitk@hijli.iitkgp.ernet.in.Abbreviations used:GAPDH,glyceraldehyde-3-phosphate dehydrogenase;MRSA,methicillin-resistant Staphylococcus aureus;SaGAPDH1,GAPDH1from Staphylococcus aureus MRSA252;TmGAPDH,GAPDH from Thermotoga maritima; BsGAPDH,GAPDH from Bacillus stearothermophilus;SaGAPDH1-P H,phosphate-bound structure of GAPDH1in holo form from Staphylococcus aureus MRSA252;SaGAPDH1-P A,phosphate-bound structure of GAPDH1in apo form from Staphylococcus aureus MRSA252;r.m.s.d.Cα,root-mean-square deviation of Cα;G3P,glyceraldehyde-3-phosphate;IntroductionStaphylococcus aureus,one of the most common causes of nosocomial infections,is responsible for a wide range of illnesses from minor skin infections to life-threatening diseases such as meningitis,pneu-monia,toxic shock syndrome,and septicemia. Amidst vast technical and medical advancements, it still continues to wreak havoc worldwide and remains one of the leading causes for morbidity and mortality.The resistance of this“golden staph”to all prevalent frontline antimicrobials has increased the menace.In fact,the notorious methicillin-resistant strain of S.aureus(MRSA)has already become an endemic in the last decade.Recent reports on the recalcitrance of this bacterium to modern glycopep-tide antibiotics such as vancomycin have increased the concern to tackle the vancomycin-resistant and vancomycin intermediate S.aureus.1,2Glycolytic enzymes,responsible for the produc-tion of ATP in the cells,are necessary for the pathogen's sustenance.The enzyme glyceralde-hyde-3-phosphate dehydrogenase(GAPDH,EC: 1.2.1.12)is the sixth enzyme of the glycolytic path-way.It acts on glyceraldehyde-3-phosphate(G3P)to convert it into1,3-bisphosphoglycerate(1,3-BPG) and consumes inorganic phosphate to harness the energy into NADH.The reaction mechanism has been intensively investigated.3–8Although the role of GAPDH as a housekeeping enzyme is well studied,recent investigations revealed new proper-ties of this enzyme.These include localization on the cell surface,binding to cellular molecules,9–14and roles in apoptosis.15GAPDHs have two anion recognition sites designated as the“P s”and the“P i”site corresponding to the binding of substrate and inorganic phosphates.The“P s”site is highly con-served in all eukaryotic and prokaryotic GAPDHs while location of“P i”site varies.16The classical“P i”site is found in Bacillus stearothermophilus while a “new P i site”was observed in Thermotoga maritima. Based on the“new P i”site,a flip-flop mechanism,in which the C3phosphate of the substrate binds to the “new P i”site and flips to the“P s”site before the hydride transfer,was proposed.17MRSA252contains two cytosolic GAPDHs—GAPDH1(National Center for Biotechnology Infor-mation accession code YP_040254)and GAPDH2 (National Center for Biotechnology Information accession code YP_041153).Because of its versatility, S.aureus is able to survive in extracellular or intracellular habitats,such as on skin or in epithelial cells,endothelial cells,and osteoblasts.In most of these environments,the resistance against reactive oxygen species might be important for survival.In 2004,Weber et al.have shown that under oxidative stress,GAPDH in S.aureus is irreversibly oxidized.18 The complete inactivation of this key enzyme by oxidation was shown to have dramatic conse-quences of the entire catabolism of the cell,as with the eukaryotic GAPDHs,there is a significant difference in the mechanism and pattern of oxida-tion between them.Thiol oxidation of SaGAPDH is completely irreversible and totally inactivates the enzyme.However,in eukaryotic GAPDHs,S-thiolation of the catalytic cysteine protects and partially inactivates the enzyme from the oxidant. This inhibition is reversible and there is a complete resumption of activity once the oxidant is removed and the enzyme is dethiolated.19Such an important enzyme such as GAPDH from S.aureus needs elaborate study from the structural and mechanistic aspect.Hence,this study targets the structural and functional investigation of GAPDH1 from S.aureus MRSA252(SaGAPDH1).In this study,different crystal structures of the wild-type enzyme in apo and holo forms,ternary complexes of active-site mutants with substrate and coenzyme(C151S.NAD.G3P,C151G.NAD. G3P,and C151S+H178N.NAD.G3P),and thioacyl intermediate have been described in detail with an aim to elucidate the mechanistic pathway.Struc-tural and functional investigations of this enzyme provide novel insights into the substrate binding and catalytic mechanism.This is the very first study to report the binding of substrate in an inverted manner.ResultsOverall structureSaGAPDH1crystallizes in P21space group con-sisting of four molecules in the asymmetric unit.The subunits,designated as O,P,Q,and R(Fig.1a),are related by three noncrystallographic2-fold axes of symmetry P,Q,and R with the choice of first monomer for denomination“O”being arbitrary.20 Each of the subunit is composed of two domains:the NAD+binding domain(residues1–150)and the catalytic domain(residues151–336)(Fig.1b).The solvent-accessible surface area of the tetrameric assembly is44,800Å.The NAD+binding domain has a classicalα/βdinucleotide binding fold—the Rossmann fold.This domain folds into nineβ-sheets comprising of residues3–8(βA),27–33(βB),58–60(βC),65–67 (βD),70–75(βE),92–95(βF),116–119(βG),128–130 (βH),and145–146(βI).The strands are inter-connected by either short loops or helices.βD and theβH runs antiparallel with the other seven parallel β-sheets.There are four helices in this domain.αB (12–23)is in betweenβA andβB whileαC comprising of residues38–46connectsβB andβC.αD(80–88) connectsβE andβF.αE(103–113)is interspersed betweenβF andβG.The catalytic domain consists of eight mixedβ-strands,β1(170–179),β2(207–210),β3(228–235),β4 (241–249),β5(271–275),β6(290–293),β7(298–302),binding domain and the catalytic domain are linked byα1.Catalytically active residues Cys151and His178reside inα1andβ1,respectively.The C-terminalα3helix(317–336)fits into a groove of the N-terminal domain and is involved in a number of interactions with the coenzyme.A prominent feature of the catalytic domain is a large S-shaped loop called“S loop”comprising of residues179–206. The flexible nature of this long unstructured region is evident from its comparatively high temperature factor values and poorly defined electron density maps for some residues in some of the subunits. Superimposition of the NAD+binding domain of the P subunit of SaGAPDH1with that of GAPDH root-mean-square deviation of Cα(r.m.s.d.Cα)of 0.64Å.Cα(1–151)was used to calculate the super-position matrix.Insignificant differences are ob-served in the helices and strands while the variations in loop regions(139–143)are noticeable.When superposed with catalytic domain of a monomer of 1HDG,r.m.s.d.Cα(152–332)of the catalytic domain of the P subunit is quite high(1.09Å).This can be accounted for a substantial variation not only in the orientation of flexible loops but also in orienta-tions of more orderedα-helices andβ-strands that occur due to insertions and deletions.The S loop is primarily responsible for the intersubunit interac-tions.The structures of SaGAPDH1presented in thisFig.1.SaGAPDH1―verall structure and coenzyme binding.(a)Spatial organization of the four subunits in the asymmetric unit:The subunits P(cyan),O(blue),Q(magenta),and R(green)are related by a noncrystallographic222 symmetry on three mutually perpendicular axes designated as P,Q,and R.P-axis is orthogonal to the plane of the paper.(b)Cartoon representation of monomeric SaGAPDH1:The N-terminal domain(colored pink)binds NAD+(shown in sticks)while the C-terminal catalytic domain(colored blue)contains the flexible long S loop.(c)Stereoview of simulated annealing omit density map(F o−F c)of NAD+contoured at3.5σ.The unbiased omit map was calculated from the refined structure before the introduction of the coenzyme.Some of the interacting residues are highlighted.Table 1.Summary of data collection and refinement statisticsHoloApoHolo_PO4(SaGAPDH1-P H )Apo_PO4(SaGAPDH1-P A )Ternary complexThioacyl complexC151S.NAD.G3P C151G.NAD.G3PC151S+H178N.NAD.G3PData collection Space group P 21Cell parameters a ,b ,c (Å)68.2,104.9,91.264.3,94.9,86.668.2,104.9,90.667.0,93.7,89.168.5,104.5,91.268.6,103.0,90.767.9,93.9,89.969.1,103.0,90.3β(°)107.7105.7107.6106.8108.0109.3107.6109.4Resolution (Å)30.78–1.70(1.76–1.70)19.65–2.50(2.63–2.50)19.14–2.50(2.59–2.50)33.34–2.20(2.28–2.20)21.35–2.50(2.59–2.20)27.28–2.20(2.28–2.20)33.83–2.60(2.69–2.60)19.91–2.80(2.94–2.80)Completeness (%)94.5(93.6)99.3(96.7)99.8(99.7)99.7(97.3)98.8(97.7)99.6(98.4)99.0(90.4)99.2(96.9)Redundancy 3.5(3.4) 3.7(3.5) 3.0(2.9) 3.6(3.3) 3.7(3.7) 3.6(3.5) 3.6(3.2) 3.8(3.7)I /σ(I )8.7(2.4)19.1(4.5)8.1(2.5) 6.8(2.3)9.7(3.2)8.3(2.5)7.4(2.4)9.8(2.7)R merge (%)a5.9(43.0)6.1(28.1)8.5(37.6)8.8(42.8)8.4(34.4)8.3(40.7)10.0(38.9)13.4(49.7)Refinement Resolution (Å)20.00–1.7020.00–2.5019.14–2.5020.00–2.2021.35–2.5027.26–2.2033.83–2.6020.00–2.80No.of reflections 126,51434,62141,92753,27941,75057,04832,75429,373R work (%)b 18.918.717.721.618.719.720.018.1R free (%)b22.124.922.425.823.824.924.824.1Average B -factors (Å2)Protein 32.226.045.852.526.323.536.923.3NAD 39.0—43.9—30.524.737.0—G3P————64.138.271.031.5Phosphate ——84.067.0————Glycerol ———————31.1Chloride ———————43.0Water 34.928.047.954.628.425.939.0625.4r.m.s.d.Bond length (Å)0.0060.0140.0150.0140.0150.0170.0140.013Bond angle (°)1.04 1.57 1.55 1.47 1.57 1.70 1.50 1.43Ramachandran plot (%)Most favored91.188.090.189.988.788.788.588.6Additionally allowed 8.711.29.910.011.110.911.211.3Generously allowed0.30.40.10.20.30.30.1Values in parentheses correspond to values in the highest-resolution shell.aR -factor for symmetry-related intensities.bR work is crystallographic R -factor.R free is calculated based on 5%of total reflections excluded from refinement.952Crystal Structure of GAPDH1from MRSAAll the subunits within each asymmetric unit are structurally similar as inferred from the r.m.s.d.Cαvalues.However,the tetramers generated by the noncrystallographic222symmetry have three nonequivalent interfaces.The P-axis interface (between subunits O and P and subunits R and Q)is the most extended interface(2058Å2)and is principally formed by theβ-strands and the S loop.The R-axis interface(between subunits O and R and subunits P and Q)is smaller(1365Å2),and finally,the Q-axis interface(between subunits O and Q and subunits R and P)extend only through 518Å2.The most extended subunit interactions are formed by the P-axis-related monomers,with58residues per subunit interacting less than 4.0Ådistance from an atom in the adjoining subunit. About40hydrogen bonds and30salt bridges between the different polar residues along the interface are observed.Coenzyme bindingNAD+was not added during the entire purifica-tion and crystallization process,but during re-strained refinement of the initial model ofholoen-zyme,a strong peak in the F o−F c omit map corresponding to10σ(Fig.1c)was observed in each of the subunit that can only be accounted for the incorporation of the coenzyme.During over-expression in Escherichia coli,the heterologous protein consumes its coenzyme from the host cells. In all other GAPDHs reported so far in the literature, the purified recombinant enzyme is principally obtained in the apo from.SaGAPDH1is the first of its kind that was overexpressed and purified with its bound coenzyme.In all the four subunits,it sits in the groove of the N-terminal domain,in an extended conformation with the nicotinamide ring pointing towards the catalytic Cys151and His178,and possesses average temperature factors similar to those of the protein.Containing polar ribose and phosphate units,the coenzyme is principally stabi-lized by salt bridges and hydrogen bonds with neighboring polar amino acid residues and solvent molecules(Supplementary Fig.1).Pi stacking interaction is observed between the aromatic side chain of Tyr320and the nicotinamide ring of NAD+ lying in parallel orientation.The C4of the nicotin-amide ring is positioned at a distance of5.89and 3.93Åfrom the imidazole NE2of the His178and SG of Cys151,respectively.His178and Cys151,along with NAD+,form the perfect pocket for the substrate binding.Phosphate binding:“P s”and“new P i”site SaGAPDH has two anion recognition sites desig-nated as“P s”and“new P i”site that correspond to the binding of the C3phosphate of G3P and inorganic phosphate required for phosphorylation. We have separately crystallized the enzyme in the presence of phosphate in both holo(SaGAPDH1-P H)and apo(SaGAPDH1-P A)forms.Analysis of the unbiased F o−F c omit maps(Fig.2a and b)of SaGAPDH1-P H and SaGAPDH1-P A clearly revealed peak of electron density above5σcorresponding to the anions.In SaGAPDH1-P H,all the“P s”and“P i”sites of the four chains contain phosphate ions with full occupancy.The“P s”site is composed of the side chains of the residues of Asp181,Thr183,and Arg234and the2′hydroxyl (O2D)of the ribose unit attached to the nicotin-amide ring of NAD+(Fig.2c).The“P i”phosphate is principally stabilized by the side chains of con-served Thr211,Ser150,His178,and Arg234and the main-chain nitrogen and side-chain hydroxyl of Thr152.The position of the“P i”phosphate is quite different from that of the“classical P i”site but similar to that found in TmGAPDH21and is generally referred to the“new P i”site.It is situated closer to the catalytic Cys151.A number of water-mediated hydrogen-bonding interactions with polar side-chain groups of amino acid residues also stabilize the phosphate ion in this new hydrophilic pocket. This“new P i”site in SaGAPDH1is3.40Åaway from the“classical P i”site obtained in GAPDH from B.stearothermophilus(BsGAPDH)(1GD1,sequence identity:50%)17(Fig.2c).The presence of phosphate ion in the“classical P i”site or in the“new P i”site depends upon the conformation of the strand–loop–helix segment containing the residues209–215in SaGAPDH1.This segment corresponds to residues 206–212in other GAPDHs where it is commonly referred to as the“206–212loop”.SaGAPDH1-P A has fully occupied“new P i”sites in the four subunits but a totally empty“P s”site(Fig. 2d).The“P s”site is occupied with water molecules. The position of“P i”phosphate is identical with that obtained in holoenzyme.An additional interaction appears in SaGAPDH1-P A between one of the oxygen atoms of the“P i”phosphate and the main-chain nitrogen of Gly212.This arises due to slight rearrangement of the209–215loop that pushes the amide nitrogen of Gly2123.30Åmore towards the “new P i”site.Structures of ternary complexes Superposition of each of the ternary complexes (C151S.NAD.G3P,C151G.NAD.G3P,and C151S+ H178N.NAD.G3P)with its corresponding binary complexes gives r.m.s.d.Cαof less than0.15Å, which proves that no drastic conformational change has been introduced upon substrate binding.Analy-sis of unbiased F o−F c maps of the three ternary complexes clearly shows a strong peak of electron density above8σin the active site that can be attributed to bound substrate.The substrate is bound in a noncovalent manner in all the three complexes.Overall B-factors of G3P are comparatively higher due to exposed active-site cavity.In C151S.NAD.G3P and C151S+H178N. NAD.G3P,G3P is bound to all the subunits withFig.2.Phosphate binding to SaGAPDH1.Stereoview of the unbiased simulated annealing(F o−F c)omit maps (contoured at3.5σ)of phosphate binding sites in(a)SaGAPDH1-P H and(b)SaGAPDH1-P A.In(a),both the“P s”and “new P i”sites are occupied by phosphate ions while in(b),only the“new P i”site is occupied by phosphate.Water molecules reside in the“P s”site.The maps were computed before the phosphate ions were introduced.(c)Phosphate ions bind to both the“P s”and the“new P i”site in all four chains of SaGAPDH1-P H.Selected stabilizing interactions of the“P s”phosphate and“new P i”phosphate with amino acid residues and solvent molecules are shown in pink and blue, respectively.The sulfate ion residing in the“classical P i”site in BsGAPDH(PDB code:1GD1)is overlaid on the structure of the SaGAPDH1-P H.The“new P i”site in SaGAPDH1-P H is3.4Åaway from the“classical P i”site in BsGAPDH.The figure shown corresponds to the Q subunit of SaGAPDH1-P H.(d)Phosphate ions bind only to the“new P i”site in all the four subunits of SaGAPDH1-P A.The“P s”site is filled by water molecules in all the four chains.Interactions of the protein atoms with the“P i”phosphate is highlighted in blue while those with water molecule in the“P s”site are shown in pink.its C3phosphate positioned in the“P s”site while the “new P i”site remains unoccupied(Fig.3a and b). This is similar to that observed in the ground-state Michaelis complexes from B.stearothermophilus but in contrast to GAPDH from Cryptosporidium parvum (PDB code:3CIF,sequence identity:46%)22where the C3phosphate is bound to the“new Pi”site in three of the four subunits and in a completely different position in the fourth.The oxygen atoms of the C3phosphate(O1P,O2P,O3P,and O4P)form extensive hydrogen bonds with the side chains of conserved Thr181,Asp183,and Arg234and with the 2′hydroxyl group of ribose(O2D)adjacent to the nicotinamide group of NAD+.The H-bonding interaction between the O1A atom of G3P and NE2of His178that is present in C151S.NAD.G3P (Fig.3a)is lost in the ternary complex of double mutant(Fig.3b).In C151G.NAD.G3P,G3P binds in a completely inverted manner with its C3phosphate positioned in the cavity lined by Ser150,Gly151,and Thr152 (Fig.3c).Placing the substrate with its C3 phosphate in the“P s”site in unbiased F o−F c map resulted in improper fitting of the ligand with some unresolved negative density over the“P s”site. However,placement of G3P in the reverse manner with the C1atom positioned in the“P s”site solved the problem.The difference electron density map of bound G3P is shown in Fig.3d.Several reasons can be put forward to explain this interesting but apparently contradictory observation.Firstly, mutation of Cys151to a glycine residue increases the size of the active-site cavity by almost10.0Å3some of the interactions between the enzyme and the ligand are compromised,some additional hydrogen-bonding interactions of oxygen atoms of C3phosphate group with the side chains and peptide backbone of the enzyme can be principally inferred to localize C3phosphate in this new orientation.The O1A,O1B,and O2in turn are hydrogen bonded to the side chains of Asp183, Arg234,and the2′hydroxyl group of the ribose (O2D)of the coenzyme(Fig.3c).In spite of using D,L-G3P in the soaking experi-ments,the C2carbon adopts an R configuration, which accounts for greater affinity of the enzyme towards D-G3P.In C151S_G3P and C151S+ H178N_G3P,the C2hydroxyl group of G3P is stabilized by hydrogen bonds with OG side chain and amide nitrogen of Ser151.Moreover,an additional stabilization may arise due to the interaction of C2–OH with a water molecule that is stabilized through hydrogen bonding to O2D and N7N of NAD+(Fig.3a and b).In the complexes C151S.NAD.G3P and C151G. NAD.G3P,G3P is bound in hydrate(gem-di-ol) form to all the four subunits.This is not unlikely because the equilibrium between the aldehyde and gem-di-ol form(Fig.4a)is strongly shifted towards the latter in solution and the proportion of the hydrate form increases with electron-with-drawing substituents.G3P has an electron-with-drawing hydroxyl and phosphate group that can stabilize the di-ol form significantly.The observa-tion is similar to that observed in the case of C149A mutant of E.coli that uses the hydrate form of 23Fig.3.Active sites of ground-state Michaelis complex:(a)C151S.NAD.G3P(Chain O):G3P is in hydrated gem-di-ol form.(b)C151S+H178N.NAD.G3P(i)Chain O:G3P is in aldehyde form.(ii)Chain Q:G3P in hydrated gem-di-ol form.(c) C151G.NAD.G3P(Chain P):G3P is in gem-di-ol form.The C3phosphate of G3P is positioned in the“P s”site of(a)and(b) while the“new P i”site remains unoccupied.G3P binds in a completely inverted manner in(c).Here,C1of G3P resides in the“P s”site.Some of the polar interactions(shown in green)of G3P with amino acid residues,NAD+,and water molecules are highlighted.(d)Stereoview of simulated annealing(F o−F c)omit map of G3P in C151G.NAD.G3P justifies the inverted orientation of substrate.The map is contoured at3.5σand is computed before the introduction of the substrate.G3P is shown in gem-di-ol form.chains in aldehyde form(O and R)(Fig.3b,i)and in gem-di-ol form to the other two(P and Q) (Fig.3b,ii).Analysis of simulated annealing F o−F c omit maps(Fig.4b and c)allows us to place the substrate unambiguously in the required form within the active-site cavity of different subunits.A careful analysis of the interactions that can principally stabilize the hydrated form with res-pect to the free aldehyde form clearly reveals the role of His178.A hydrogen bond between O1A of G3P and NE2of His178at a distance of2.65Åin C151S.NAD.G3P is one such stabilizing interaction that is lost in the ternary complex of the double mutant.Absence of such an important interaction may be postulated to shift the equilibrium more towards the aldehyde form.Some subtle differ-ences between the structures of SaGAPDH1and BsGAPDH24may be responsible for the substrate to exist in two different forms in these two enzymes that however remain inexplicablebecauseFig.4.Views of D-G3P molecule.(a)Equilibrium between the aldehyde and hydrate(gem-di-ol)form of D-G3P.The equilibrium is strongly favored towards the hydrated form in aqueous solution owing to extensive hydrogen bonding.C2 is chiral and is in“R”configuration.O1and O2point in the opposite direction.The atomic numbering of D-G3P used in this article is depicted here.Stereoview of simulated annealing F o−F c map of D-G3P in(b)aldehyde form and(c)gem-di-ol (hydrate)forms found in C151S+H178N.NAD.G3P.The unbiased difference maps are calculated before the introductionthe active-site residues and geometry are more or less conserved.The absence of a covalent bond formation between C1of D-G3P and Ser151is justified from a kinetic point of view.The rate of acylation is decreased by23,000times when the active-site Cys151is mutated to serine.In the crystallization medium containing polyethylene glycol(PEG) 4000,the acylation rate is probably even more retarded than in aqueous solution,which helps the noncovalently bound G3P to be stable in the ternary complex even after soaking with50mM G3P for10min.Structure of thioacyl intermediateThe mechanism of oxidative phosphorylation proceeds via a thioacyl intermediate.We were able to isolate the intermediate in the case of apoenzyme,but it was not possible to trap such an intermediate in the case of holoenzyme as the reaction was instantaneous.G3P binds to the catalytic site in all the four chains.Unbiased difference map of the thioacyl intermediate is shown in Fig.5a.Although noncrystallographic restraints were not imposed on the substrate,it essentially binds to all the four monomers with identical conformations sharing almost similar interactions.The thioester bond is formed between the sp2hybridized C1of G3P and SG of catalytic Cys151(Fig.5b).The phosphate group is now positioned in the“new P i”site in contrast to the “P s”site as found in the ternary complexes of the mutant proteins.The“new P i”site is positioned at a distance of 6.0Åfrom the“P s”site.Phosphate group in the“new P i”site is stabilized by a number of hydrogen-bonding interactions with side-chain and main-chain atoms of conserved His178and Thr211,respectively,and guanidium group of Arg234via a water molecule.This shift of the C3P group from“P s”to“new P i”site is associated with a significant rearrangement of the carbon chain of the ligand.While O1is oriented towards His178in the ternary complexes,it points away from catalytic histidine in the thioacyl intermediate(Fig.5c).It is observed that the C2-O2bond in the thioacyl intermediate is parallel with the plane of the nicotinamide ring of the coenzyme but is oriented perpendicularly in the ternary complex.The preferential uptake of the D isomer by the enzyme can be explained from the structure of the thioacyl complex.In the case of D-G3P,the intermedi-ate clearly shows that C1and C2of the phosphogly-cerol moiety both adopt an R configuration that places O1atom trans to O2(Fig.5a).In the case of L-G3P,the S configuration of C2orients O1and O2cis to each other.The two hydroxylic groups then are sufficiently close to make an H-bonding interaction that can stabilize the cis conformation,but the Pitzer(torsional) strain developed due to their dihedral angle makes the overall interaction unfavorable.The lower energy associated with trans conformation explains the higher affinity of the enzyme towards D-G3P. Conformations of the209–215loop in comparison to other GAPDH structuresIn all the structures of SaGAPDH1discussed,the 209–215loop exists in two conformations—Confor-mation I and Conformation II(Fig.6a).Conformation Fig.4(legend on previous page)。
AhR信号通路与HIF-1信号通路的相互作用
M OLECULAR AND C ELLULAR B IOLOGY,July2009,p.3465–3477Vol.29,No.13 0270-7306/09/$08.00ϩ0doi:10.1128/MCB.00206-09Copyright©2009,American Society for Microbiology.All Rights Reserved.The Active Form of Human Aryl Hydrocarbon Receptor(AHR)Repressor Lacks Exon8,and Its Pro185and Ala185Variants Repressboth AHR and Hypoxia-Inducible Factorᰔ†Sibel I.Karchner,1Matthew J.Jenny,1Ann M.Tarrant,1Brad R.Evans,1,2Hyo Jin Kang,3Insoo Bae,3David H.Sherr,4and Mark E.Hahn1*Biology Department,Woods Hole Oceanographic Institution,Woods Hole,Massachusetts025431;Biology Department, Boston University,Boston,Massachusetts022152;Department of Oncology,Georgetown University Medical Center, Washington,DC200073;and Department of Environmental Health,Boston University School ofPublic Health,Boston,Massachusetts021184Received16February2009/Accepted10April2009The aryl hydrocarbon receptor(AHR)repressor(AHRR)inhibits AHR-mediated transcription and has beenassociated with reproductive dysfunction and tumorigenesis in humans.Previous studies have characterizedthe repressor function of AHRRs from mice andfish,but the human AHRR ortholog(AHRR715)appeared tobe nonfunctional in vitro.Here,we report a novel human AHRR cDNA(AHRR⌬8)that lacks exon8ofAHRR715.AHRR⌬8was the predominant AHRR form expressed in human tissues and cell lines.AHRR⌬8effectively repressed AHR-dependent transactivation,whereas AHRR715was much less active.Similarly,AHRR⌬8,but not AHRR715,formed a complex with AHR nuclear translocator(ARNT).Repression of AHR byAHRR⌬8was not relieved by overexpression of ARNT or AHR coactivators,suggesting that competition for these cofactors is not the mechanism of repression.AHRR⌬8interacted weakly with AHR but did not inhibit its nuclear translocation.In a survey of transcription factor specificity,AHRR⌬8did not repress the nuclear receptor pregnane X receptor or estrogen receptor␣but did repress hypoxia-inducible factor(HIF)-dependent signaling.AHRR⌬8-Pro185and-Ala185variants,which have been linked to human reproductive disorders,both were capable of repressing AHR or HIF.Together,these results identify AHRR⌬8as the active form of human AHRR and reveal novel aspects of its function and specificity as a repressor.The aryl hydrocarbon receptor(AHR)repressor(AHRR)is a basic-helix-loop-helix/Per-AHR nuclear translocator(ARNT)-Sim(bHLH-PAS)protein discovered because of its similarity to the AHR,a ligand-activated transcription factor involved in the response to synthetic aromatic hydrocarbons(48).The AHR and AHRR form a negative regulatory loop that is evolutionarily conserved in vertebrates(32);expression of AHRR is regulated by the AHR,and AHRR acts as a transcriptional repressor of AHR function(1,32,48).Like the AHR,AHRR can dimerize with the ARNT,and the AHRR-ARNT complex can bind to AHR-responsive enhancer elements(AHREs).Repression oc-curs through competition between AHR and AHRR for binding to AHREs(14,48)as well as through additional mechanisms that do not involve competition for ARNT and are independent of AHRE binding by AHRR(14).The biological and toxicological functions of AHRR are not well understood,but recentfindings suggest that AHRR is involved in human reproductive physiology and in the regula-tion of cell growth(reviewed in references20and22).A human AHRR(hAHRR)Ala185Pro polymorphism has been associated with altered reproductive development and infertil-ity in men(16,46,59,64)and endometriosis in women(19,35,62,65),but the functional properties of the polymorphic vari-ants have never been assessed.AHRR overexpression inhibits the growth of human tumor cells in culture(30,56,68).Con-versely,knockdown of AHRR expression enhances cell growth and confers resistance to apoptosis;consistent with this,the AHRR gene has been found to be silenced by hypermethylation in a variety of human cancers(71).Based on these and other findings,the AHRR has been proposed to function as a tumor suppressor gene(22,71).In order to assess the functions of AHRR and its polymor-phic variants and their relationship to human disease,it is important to understand the nature of the transcripts and proteins encoded by the AHRR gene,as well as their expres-sion in human tissues and cell lines.An hAHRR cDNA iden-tified in a large-scale screen of cDNAs from brain(50)encodes a protein of715amino acids(aa)(referred to here as AHRR715).The human AHRR gene encoding this protein has been reported to contain12exons,thefirst of which is non-coding(8,16).Our initial functional analysis of this protein suggested that,unlike AHRRs from mouse,frog,andfish(15,32,48,70),human AHRR715was not an effective repressor of AHR function in vitro.In phylogenetic analyses involving amino acid sequence alignments of multiple vertebrateAHRRs,we identified an18-aa segment of AHRR715that was absent from all other AHRRs.We therefore hypothesized the existence of an alternative hAHRR form lacking this segment and also hypothesized that this alternative form might exhibit characteristic repressor function.*Corresponding author.Mailing address:Biology Department,MS#32,Woods Hole Oceanographic Institution Woods Hole,MA 02543.Phone:(508)289-3242.Fax:(508)457-2134.E-mail:mhahn@.†Supplemental material for this article may be found at http://mcb/.ᰔPublished ahead of print on20April2009.3465Here,we report the identification and cloning of a novel hAHRR cDNA that lacks exon8of the original AHRR clone. This new AHRR(AHRR⌬8)is the predominant form ex-pressed in multiple human tissues and human tumor cell lines. We compare the functions of the two AHRR splice variants and provide thefirst functional mechanistic assessment of the hAHRR Pro185Ala polymorphic variants that have been as-sociated with increased susceptibility to reproductive dysfunc-tion in human populations.We also show that competition for ARNT or AHR coactivators is not involved in the mechanism of AHR repression and that human AHRR⌬8(hAHRR⌬8)is capable of repressing hypoxia-inducible factor(HIF)-depen-dent signaling.MATERIALS AND METHODSChemicals and cell lines.2,3,7,8-Tetrachlorodibenzo-p-dioxin(TCDD)was obtained from Ultra Scientific(Hope,RI).Dimethyl sulfoxide(DMSO),clo-trimazole,cobalt chloride,and17-estradiol were obtained from Sigma-Aldrich (St.Louis,MO).COS-7cells and the human cell lines HepG2,HeLa,MCF-7, Hs578T,and MDA231were obtained from the American Type Culture Collec-tion(ATCC;Manassas,VA)and grown according to standard procedures.BP-1 cells were generously provided by J.Russo(Fox Chase Cancer Center,Phila-delphia,PA).Human cDNA panel screening.The expression of AHRR exon8in various human tissues was determined by amplifying a partial fragment of AHRR with primersflanking the exon8region.Primers hRR-F635(5Ј-AGTACTCGGCCT TCCTGACC-3Ј)and hRR-R816(5Ј-CGCCTTCTTCTTCTGTCCAA-3Ј)were used with5l of cDNAs from adult and fetal human cDNA panels(BD Biosciences,Mountain View,CA)in a25-l amplification reaction mixture using AmpliTaqGold DNA polymerase(Applied Biosystems,Foster City,CA).The PCR conditions were as follows:94°C for10min,94°C for15s and60°C for30s for35cycles,and72°C for7min.The PCR products were resolved on15% Tris-borate EDTA gels.Screening of human cell lines for the Pro/Ala polymorphism and the presence of exon8.Total RNA was isolated from human cell lines MCF-7,Hs578T, MDA231,and BP-1as described earlier(68).Briefly,cells were frozen and pulverized into afine powder.Total cellular RNA was isolated using RNAzol as described by the manufacturer(Leedo Medical Laboratories,Houston,TX). RNA was quantified with a spectrophotometer at optical densities of260nm and 280nm.cDNA was synthesized from2g of total RNA using Omniscript reverse transcriptase(Qiagen,Valencia,CA).A partial fragment of AHRR was ampli-fied from cDNA derived from each cell line using primers hRR-F494(5Ј-AGG ACTTCTGCCGGCAGCTCC-3Ј)and RRex8R(5Ј-CAGCTGCCAAGCCTGT GACC-3Ј)flanking the region containing the Pro185Ala polymorphism and exon 8.The PCR products were cloned into the pGEM-T vector(Promega),and multiple clones were sequenced for each cell line.Generation of AHRR plasmid constructs.The pcDNAhAHRR(AHRR715) construct was prepared by subcloning the KIAA1234cDNA(clone fH08618;a gift from Takahiro Nagase,Kazusa DNA Research Institute,Chiba,Japan[50]) into pcDNA3.1,as we described earlier(32).Full-length hAHRR⌬8was ampli-fied from testes cDNA(human cDNA panel;BD Biosciences,Mountain View, CA)using primers hRR-F39(5Ј-GATCATATGCCGAGGACGAT-3Ј)and hRR-R2227(5Ј-GAGCTTGGATGGTGGTCACT-3Ј)and Advantage DNA polymerase(BD Biosciences).The PCR conditions were as follows:94°C for1 min;94°C for5s,64°C for10s,and68°C for2.5min forfive cycles;94°C for5s, 62°C for10s,and68°C for2.5min forfive cycles;94°C for5s,60°C for10s,and 68°C for2.5min for25cycles;72°C for10min.The amplified product was cloned into the pGEM-T vector(Promega,Madison,WI)and sequenced.The insert was cut out of EcoRI and SpeI sites and transferred to the EcoRI and XbaI sites of the pcDNA3.1vector(Invitrogen,Carlsbad,CA).Other plasmid constructs.The pEF-hAHR and mouse AHR-yellowfluores-cent protein(YFP)fusion constructs were provided by Gary Perdew(Pennsyl-vania State University,University Park,PA).The plasmid pGudLuc6.1,which contains thefirefly luciferase reporter under the control of a mouse mammary tumor virus promoter regulated by four AHREs from the murine CYP1A1 promoter,was a gift from M.Denison(University of California,Davis,CA).Rat Cyp1a1-Luc and human XRE.1A1-Luc were obtained from R.Barouki(Univer-sity of Rene Descartes,France)and S.K.Kim(Seoul National University,South Korea),respectively.Expression constructs for human ARNT and the hypoxia-responsive luciferase reporter,HRE-luc(PL949[25]),were obtained from C. Bradfield(University of Wisconsin,Madison,WI).The human pregnane X receptor(PXR)expression construct was provided by S.Kliewer(University of North Carolina at Chapel Hill),and the PXR reporter construct(XREM-tk-luc) was obtained from J.Moore(Molecular Discovery Research,GlaxoSmithKline, Research Triangle Park,NC).The human estrogen receptor␣(ER␣)construct and an estrogen-responsive luciferase reporter(3xERE-TATA-luc)were ob-tained from D.McDonnell(Duke University Medical Center,Durham,NC). Expression constructs for the receptor coactivators GRIP,CoCoA,and GAC63 were provided by M.Stallcup(University of Southern California,Los Angeles, CA).Src-1a and Src-1e constructs were obtained from E.Kalkhoven(University Medical Center Utrecht,The Netherlands)and M.Parker(Imperial College London,United Kingdom).The p300expression construct was from Upstate Biotechnologies(Lake Placid,NY).Transient transfections and luciferase assays.Transient transfections were performed as described earlier(14,32).Briefly,transfections of DNA with Lipofectamine2000reagent(Invitrogen,Carlsbad,CA)were carried out in triplicate wells24h after plating.Approximately300ng of DNA was complexed with1l of Lipofectamine2000and then added to cells;the amount of DNA used for each expression construct is listed in thefigure legends.The total amount of DNA was kept constant by adding in empty vector.Five hours after transfection,cells were exposed to DMSO(0.5%),TCDD(10nMfinal concen-tration),clotrimazole(10Mfinal concentration),CoCl(150Mfinal concen-tration),or17-estradiol(10nMfinal concentration).(For transfections involv-ing17-estradiol and ER␣,cells were grown in phenol red-free medium with charcoal-stripped serum.)Renilla luciferase(pRL-TK or pGL4.74;Promega, Madison,WI)was used as the transfection control.Cells were lysed19h after dosing,and luminescence was measured using the dual luciferase assay kit (Promega)in a TD20/20luminometer(Turner Designs,Sunnyvale,CA).The final values are expressed as a ratio of thefirefly luciferase units to the Renilla luciferase units.Experiments were repeated multiple times.AHRR antibody production and Western blots.Polyclonal antibodies to hAHRR(designed to recognize both forms)were raised in two rabbits(21st Century Biochemicals,Marlboro,MA)by coimmunizing the animals with two peptides corresponding to amino acid residues18to31(LQKQRPAVGAE KSN)and80to101(FQVVQEQSSRQPAAGAPSPGDS).To avoid cross-re-activity with AHR,the peptides were in regions of the AHRR protein exhibiting low sequence identity with the AHR(see Fig.S1in the supplemental material). Preimmune serum and serum from six bleeds were collected over the course of several weeks,and antibody titer was tested by Western blotting using hAHRR-transfected COS-7cell lysates;lysates from COS-7cells transfected with empty vector served as a control for specificity.COS-7cells were plated and transfected as described above.Twenty-four hours after transfection,cells were rinsed with phosphate-buffered saline and resuspended in2ϫsample treatment buffer.Cell lysates were subjected to sodium dodecyl sulfate-polyacrylamide gel electro-phoresis,and the gels were blotted onto nitrocellulose.The serum antibody titer for each bleed was tested by blotting with two dilutions(1:250and1:1,000).Two affinity-purified polyclonal antibodies were isolated from serum(bleeds3thru6) from a single rabbit by separate affinity purification procedures using the two individual peptides.The affinity-purified antibodies are designated PAb-RR-18-1 (against residues18to31)and PAb-RR-80-2(against residues80to101).The specificity of the affinity-purified polyclonal antibodies was assessed by blotting against lysates from COS-7cells transfected with plasmids for hAHRR,human AHR,mouse AHRR,and killifish AHRR.All results reported here(Western blots and immunoprecipitations)were performed using PAb-RR-80-2. Expression of hAHRR protein in the transient-transfection assays was mea-sured by Western blotting with PAb-RR-80-2(3g/ml),followed by a goat anti-rabbit immunoglobulin G(IgG)Ϫhorseradish peroxidase(Upstate/Milli-pore,Billerica,MA)secondary antibody(1:5,000).The AHRR proteins were then visualized by enhanced chemiluminescence(ECL-Plus;GE Healthcare, Piscataway,NJ).Coimmunoprecipitation assay.The full-length AHRR715,AHRR⌬8,AHR, and ARNT proteins were synthesized by in vitro transcription and translation (TnT;Promega,Madison,WI)in the presence or absence of[35S]methionine (MP Biomedicals,Solon,OH).Five microliters of unlabeled protein was mixed with15l of radiolabeled protein and incubated at room temperature for2h. For mixtures containing AHR,TCDD(10nM)was added.The mixtures were adjusted to25mM HEPES,200mM NaCl,1.2mM EDTA,10%glycerol,and 0.1%Nonidet P-40,pH7.4,with protease inhibitors(immunoprecipitation buffer).After two rounds of preclearing with normal mouse IgG and protein G-agarose,5g of specific antibody or IgG was added and incubated for2h, followed by precipitation with protein G-agarose overnight.The beads were washed two times with IP buffer,boiled in sample treatment buffer,and subjected3466KARCHNER ET AL.M OL.C ELL.B IOL.to sodium dodecyl sulfate-polyacrylamide gel electrophoresis on an8%poly-acrylamide gel.The gels were dried and visualized byfluorography.hAHRR antibody(PAb-RR-80-2)or normal rabbit IgG was used to precipitate AHRR complexes and nonspecific complexes,respectively.For ARNT complexes, monoclonal ARNT antibody(MA1-515;Affinity BioReagents,Golden,CO)and normal mouse IgG were used.Subcellular localization of mouse AHR-YFP.COS-7cells were grown on coverslips in six-well plates.Cells were cotransfected with350ng of mouse AHR-YFP and350ng of human ARNT expression constructs,with or without 350ng of hAHRR⌬8construct,using Lipofectamine2000reagent(Invitrogen). Luciferase reporter pGudLuc6.1(300ng)and the transfection control pRL-TK (40ng)were also transfected.Cells were dosed with DMSO or TCDD(10nM final concentration)5h after transfection.Twenty-four hours after transfection, cells were washed with1ϫphosphate-buffered saline andfixed in4%formalde-hyde.The coverslips were inverted onto slides and mounted with Vectashield hard-setting mounting medium(Vector Laboratories,Burlingame,CA).Cells were visualized using a Zeiss Axio Imager.Z1fluorescence microscope,and Axiovision software was used to collect the images.To confirm the effectiveness of AHRR⌬8as a repressor under the conditions of the assay,luciferase was measured in a plate of cells run in parallel.Nucleotide sequence accession numbers.The AHRR⌬8sequences have been deposited in GenBank,with accession numbers EU293605(mRNA)and ABX89616(protein).RESULTSIdentification and characterization of the major form of hAHRR.In our earlier studies of the evolutionary conservation of AHRR in vertebrates(15,32),we noticed that the predicted 715-aa protein derived from the original hAHRR cDNA(AHRR715[50])included an18-aa segment that had no coun-terpart(homologous amino acids)in other AHRRs.A more recent phylogenetic analysis involving an alignment of all known(i.e.,verified)mammalian,amphibian,andfish AHRR protein sequences confirmed that this unique18-aa segment ispresent only in the human AHRR715(Fig.1A).This segmentis located in an otherwise conserved portion of the PAS region downstream of the PAS-A repeat(sometimes referred to as the“intervening region”[10]between PAS repeats)(see Fig. S1in the supplemental material);it is encoded by a single exon of54nucleotides in the human genome(Fig.1B),correspond-ing to exon8described by others(8,16).AHRR715,the715-aaprotein encoded by the cDNA containing this exon,functioned poorly as a repressor in transient-transfection assays in three different laboratories(unpublished results;see below).We therefore hypothesized that there might be an alternatively spliced AHRR transcript lacking this exon and that the protein encoded by this alternative transcript might have a repressor function like that of AHRRs from other species.Using primersflanking exon8,we used PCR to amplify this region of the AHRR transcript from human tissue cDNAs. The primary amplicon was128bp,the size predicted for a form lacking exon8,rather than the182bp predicted for the exon 8-containing transcript.Subsequently,we amplified,cloned, and sequenced a full-length cDNA of2,173bp with an open reading frame of2091bp encoding a predicted AHRR proteinof697aa.The new cDNA is identical to AHRR715(50),exceptthat it lacks the sequences corresponding to exon8and thus has been designated AHRR⌬8.Two polymorphic variants of AHRR⌬8,corresponding to the Ala185Pro single nucleotide polymorphism described earlier(65),were found among the sequenced clones.To assess the relative expression of AHRR715andAHRR⌬8,we performed PCR analysis on cDNA from human tissues with primersflanking exon8,designed to produce am-plicons of different sizes for the two AHRR variants.A survey of multiple adult and fetal tissues demonstrated that AHRR⌬8 is the predominant,and in most cases only,form of AHRR expressed(Fig.1C).We also examined the relative expression of AHRR715and AHRR⌬8and the presence of Ala185Pro polymorphic variants in several human tumor cell lines.As seen with the human tissues,AHRR⌬8was the predominant form of AHRR expressed in HeLa,HepG2,MCF-7,Hs578T, MDA231,and BP-1cells(see Fig.S2A and Table S1in thesupplemental material).Although AHRR715transcripts were not detected by using primersflanking exon8,use of a primerwithin exon8showed that AHRR715transcripts were ex-pressed at low levels in HeLa and HepG2cells(see Fig.S2B in the supplemental material).Sequencing of AHRR cDNA clones from each of the cell lines confirmed AHRR⌬8as the predominant expressed form and showed that all of the cell lines except BP-1are heterozygous for the Ala185Pro poly-morphism(see Table S1in the supplemental material). AHRR⌬8and AHRR differ in repressor activity.To com-pare the functional properties of the original715-aa AHRRprotein(AHRR715)and AHRR⌬8,we performed transient-transfection assays in which we measured the ability of the AHRRs to inhibit the TCDD-inducible transactivation of re-porter gene construct pGudLuc6.1mediated by either trans-fected or endogenously expressed AHR.After transfectionwith the respective constructs,AHRR715and AHRR⌬8pro-teins were expressed at similar levels in COS-7cells,as as-sessed by Western blotting using an hAHRR antibody that recognizes both AHRR forms but does not recognize human AHR(Fig.2A and B).When transfected into COS-7cells with ARNT in the presence or absence of TCDD,neither AHRR form was able to activate transcription of pGudLuc6.1(see Fig. S3A in the supplemental material).Thus,hAHRRs lack a function as transcriptional activators,as observed previously for AHRRs from other species(15,32,48).To test the ability of AHRR715and AHRR⌬8to repress AHR-mediated signaling,each form was transfected into COS-7cells together with expression constructs for human AHR and ARNT and pGudLuc6.1.In the absence of cotrans-fected AHRR,AHR and ARNT caused transactivation of the luciferase reporter that was enhanced by TCDD(Fig.2C).Transfection of the AHRR715expression construct at50and 150ng/well caused little change in the AHR-dependent acti-vation of luciferase expression or its induction by TCDD.In contrast,AHRR⌬8at50or150ng/well completely repressed both constitutive(i.e.,exogenous ligand-independent)and TCDD-inducible reporter gene activity(Fig.2C).Similarly,AHRR⌬8,but not AHRR715,repressed transactivation by mouse AHR in COS-7cells(see Fig.S3B in the supplemental material).To evaluate the effect of the AHRRs on endog-enously expressed human AHR,AHRR715or AHRR⌬8was cotransfected with pGudLuc6.1into HepG2cells,which ex-press abundant AHR(13).As we saw with the transfectedAHRs in COS-7cells,AHRR715was ineffective as a repressor of the endogenous HepG2AHR,whereas AHRR⌬8reduced TCDD-inducible reporter gene activation by61%(Fig.2D).AHRR⌬8also was much more effective than AHRR715at repressing endogenous AHR in MCF-7cells cotransfected with two different reporter gene constructs(Cyp1a1-Luc andV OL.29,2009HUMAN AHRR⌬8AND VARIANTS REPRESS AHR AND HIF3467XRE.1A1-Luc)(Fig.2E;see also Fig.S3C in the supplemental material).(The lower percent repression in HepG2and MCF-7cells than in COS-7cells likely reflects the fact that endogenous AHR is expressed in all HepG2and MCF-7cells,whereas not all of the cells take up transiently transfected AHRR.)Together,these results demonstrate that the much greater ability of AHRR ⌬8than of AHRR 715to repress AHR transactivation is consistently observed for transfected and en-dogenously expressed AHRs from humans and mice,in three different cell lines,and with AHRE-containing reporter gene constructs derived from three different mammalian species.AHRR ⌬8and AHRR 715differ in ability to interact with ARNT.The 18-aa peptide encoded by exon 8in AHRR 715lies in the ϳ100-aa “intervening region”downstream of the PAS-A repeat.This region is highly conserved in AHR and AHRR proteins (Fig.1A;see also Fig.S1in the supplemental mate-rial),and in the AHR,it has been shown to be important for dimerization with ARNT (10,45).This region also appears to be important for dimerization of AHRR and ARNT,because deletion of this region in the zebrafish AHRRa caused a dra-matic reduction in the ability of AHRRa to interact with ARNT (14).To determine whether the presence of the 18-aa peptide within the intervening region of AHRR 715affects its ability to dimerize with ARNT,we performed a coimmuno-precipitation experiment in which radiolabeled ARNT was in-cubated with in vitro-expressed AHRR ⌬8or AHRR 715and the complex was immunoprecipitated with affinity-purified antibody against hAHRR,which recognizes both forms.AHRR ⌬8and AHRR 715were synthesized to similar levels by in vitro transcription and translation (Fig.3A).AHRR ⌬8FIG.1.Identification of hAHRR splice variant AHRR ⌬8as the major variant expressed in human tissues.(A)Partial amino acid sequence alignment of AHRRs and AHRs from different species.The top sequence is human AHRR 715;immediately below it is the sequence of AHRR ⌬8.Abbreviations:Hs,human;Mm,mouse;Rn,rat;Xl,frog;Fh,killifish;Tr,Japanese puffer fish;Dr,zebrafish;Mt,tomcod.For GenBank accession numbers,see the legend to Fig.S1in the supplemental material.(B)hAHRR gene structure.Translated exons are shown in black boxes.The first exon and part of the second exon are untranslated.The additional exon (exon 8)is shown in gray.The position of the Pro185Ala polymorphism is marked by an arrow.(C)Survey of adult human tissues for the expression of AHRR 715and AHRR ⌬8transcripts.A partial AHRR cDNA fragment was amplified from adult and fetal human cDNAs,using primers flanking exon 8(shown in gray in panel B).The presence of the exon would have produced a 182-bp PCR product (AHRR 715),as opposed to the 128-bp product (AHRR ⌬8)obtained in all tissues.3468KARCHNER ET AL.M OL .C ELL .B IOL .formed a complex with ARNT that could be specifically and strongly immunoprecipitated by the AHRR antibody (Fig.3B,lane 3versus lane 4;see also Fig.S4B in the supplemental material).In contrast,AHRR 715did not interact with ARNT (Fig.3B,lane 1versus lane 2;see also Fig.S4B in the supple-mental material).The results suggest that the presence of the 18-aa peptide encoded by exon 8disrupts ARNT dimerization,that hAHRR requires ARNT to repress AHR,and that the difference in the repressor function of AHRR ⌬8and AHRR 715reflects the inability of the latter protein to associate with ARNT.Functional comparison of AHRR ⌬8-Ala 185and AHRR ⌬8-Pro 185polymorphic variants.An Ala185Pro polymorphism in the hAHRR has been associated with human diseases in sev-eral studies (16,19,35,46,59,62,64,65),but the functional properties of the two variants have never been assessed.Both of these polymorphic variants were present in our pool of AHRR ⌬8clones.To compare their abilities to repress AHR transactivation,we performed transient-transfection assays in which we measured the abilities of AHRR ⌬8-Ala 185and AHRR ⌬8-Pro 185to repress AHR-and ARNT-dependent transactivation of pGudLuc6.1in COS-7cells.The AHRR ⌬8variants were expressed at similar levels in the transfected cells (Fig.4A).Both AHRR ⌬8-Ala 185and AHRR ⌬8-Pro 185were effective at repressing constitutive (exogenous ligand-indepen-dent)and TCDD-inducible expression of pGudLuc6.1.In ex-periments in which increasing amounts of AHRR ⌬8expres-sion constructs were transfected,the two variants were equally potent at repressing AHR-mediated transactivation of the re-porter gene (Fig.4B and C).We conclude that both AHRR ⌬8-Ala 185and AHRR ⌬8-Pro 185are fully functional as repressors of AHR and thus that the Ala185Pro polymorphism does not affect repression of AHR-mediated transcription.Mechanism of repression of AHR by AHRR ⌬8.Mimura et al.(48)showed that mouse AHRR could dimerize with ARNT and bind to AHREs and proposed that the mechanism of repression involved competition between AHR and AHRR for binding to ARNT and for binding to AHRE sequences.Our recent studies using the zebrafish AHRRa provided evidence that competition for ARNT is not an important element of the mechanism of repression and that AHRE binding may con-tribute to the repression but is not required (14).To assess the role of competition for ARNT in the repression of human AHR by hAHRR ⌬8,we performed a series oftransient-trans-FIG.2.Repressor activity of hAHRR splice variants AHRR 715and AHRR ⌬8.(A)The hAHRR antibody does not recognize human AHR or AHRRs from other species.Cell lysates from COS-7cells transiently expressing the indicated constructs were blotted and probed with antibody PAb-RR-80-2raised against the hAHRR.(B)In transient-transfection assays in COS-7cells,AHRR 715and AHRR ⌬8are expressed at similar levels.Cell lysates were blotted and probed with the hAHRR antibody.Numbers at left of panels A and B are molecular masses in kilodaltons.(C)Repression of exogenously expressed AHR by AHRR 715and AHRR ⌬8in COS-7cells.COS-7cells were transfected with human AHR (5ng),human ARNT (25ng),and AHRR 715or AHRR ⌬8constructs (50or 150ng each),along with a luciferase reporter under the control of dioxin response elements (pGudLuc6.1)and a transfection control plasmid expressing Renilla luciferase (pRL-TK).Cells were dosed with DMSO or TCDD (10nM final concentration),followed by a luciferase assay.The results shown are representative of seven independent experiments.(D and E)Repression of endogenous AHR in HepG2(D)and MCF-7(E)cells by AHRR 715and AHRR ⌬8.Cells were transfected with AHRR 715or AHRR ⌬8constructs (25and 100ng each),along with a luciferase reporter under the control of dioxin response elements (for HepG2,pGudLuc6.1;for MCF-7,Cyp1a1-Luc)and pRL-TK.Cells were dosed with DMSO or TCDD (10nM final concentration),followed by a luciferase assay.The results shown in panels D and E are representative of two and three independent experiments,respectively.V OL .29,2009HUMAN AHRR ⌬8AND VARIANTS REPRESS AHR AND HIF 3469。
巨噬细胞极化对动脉粥样硬化发生发展的影响
·综述·巨噬细胞极化对动脉粥样硬化发生发展的影响王立1a,2,陈玉辉1a,2,徐鸿轩1b,2,孟令丙1b,3,刘德平1b,2,3,龚涛1a,2,31.北京医院,a神经内科,b心血管内科国家老年医学中心中国医学科学院老年医学研究院,北京100730;2.国家老年医学中心国家卫生健康委员会北京老年医学研究所;3.中国医学科学院北京协和医学院研究生院[摘要] 动脉粥样硬化的发生和发展主要由局部血管壁炎症和脂质积累引发。
巨噬细胞在病灶演变中发挥着核心作用。
在动脉粥样硬化病变中,巨噬细胞受到各种各样的微环境信号调节,如细胞因子、氧化脂质、亚铁血红素等,从而导致其表型极化。
目前已证实巨噬细胞表型谱主要由T辅助细胞1(Th 1)细胞因子诱导的M1型和由Th 2细胞因子诱导的M2型为主,其中M2型又细分为M2a、M2b、M2c、M2d,此外尚有Mox型、M4型、M(Hb)和Mhem型。
对巨噬细胞表型可塑性机制的研究成为制定抑制或稳定动脉粥样硬化斑块治疗策略的重要内容。
近年来,社会心理因素在动脉粥样硬化发生发展中的作用已成为研究的热点。
慢性心理应激时交感肾上腺髓质(SAM)系统和下丘脑-垂体-肾上腺轴(HPA)的持续激活导致儿茶酚胺类激素和糖皮质激素分泌显著增加,它们对巨噬细胞表型可塑性的调节和功能后果产生重要影响。
该文对动脉粥样硬化斑块中巨噬细胞亚群的特征,及在多种组织中神经内分泌激素如儿茶酚胺类激素和糖皮质激素对巨噬细胞表型可塑性的影响进行综述。
[关键词] 动脉粥样硬化;巨噬细胞;糖皮质激素类;儿茶酚胺类;应激,心理学;综述DOI:10.3969/J.issn.1672 6790.2022.01.032EffectofmacrophagepolarizationontheinitiationandprogressionofatherosclerosisWangLi ,ChenYuhui,XuHongxuan,MengLingbing,LiuDeping,GongTaoDepartmentofNeurology,BeijingHospital,NationalCenterofGerontology;InstituteofGeriatricsMedicine,ChineseAcademyofMedicalSciences,Beijing100730,ChinaCorrespondingauthor:GongTao,Email:gb20598@sina.com[Abstract] Theinitiationandprogressionofatherosclerosisaremainlycausedbyinflammationandlipidaccumulationinthelocalvascular.Macrophagesplayacentralroleinthedevelopmentandprogressionoflesions.Intheatheroscleroticlesions,macrophagesareregulatedbyavarietyofmicroenvironmentalfactors,suchascytokines,oxidizedlipids,andhemeiron,whicheffectthephenotypicpolarization.Nowadays,ithasbeenconfirmedthatthemacrophagephenotypespectrumischaracterizedmainlyclassicalM1macrophagesinducedbyT helper1(Th 1)cytokinesandbythealternativeM2macrophagesinducedbyTh 2cytokines,ofwhichM2macrophagescanbefurtherclassifiedintoM2a,M2b,M2candM2dsubtypes.Andatheroscleroticplaque specificmacrophagephenotypesincludeM4type,M(Hb)orMhemtype,Moxtype.Studyingthemechanismofmacrophagephenotypicplasticityhasbecomeanimportantworktoexploitnewwaytoinhibitandstabilizeatherosclerosis.Inrecentyears,theeffectsofsocialpsychologicalfactorsonthedevelopmentofAShavebecomeahottopic.Thecontinuousactivationofthesympatheticadrenalmedulla(SAM)systemandthehypothalamic pituitary adrenalaxis(HPA)duringpsychosocialstressleadstoasignificantincreaseinthesecretionofcatecholaminesandglucocorticoids,whichplayanimportantroleintheregulationofmacrophagephenotypicplasticityanditsfunctionalconsequences.Thisreviewsummarizestheknowledgeofmacrophagesubsetsinatheroscleroticplaquesandtheinfluenceofneuroendocrinehormonessuchascatecholaminesandglucocorticoidsonthephenotypicplasticityofmacrophagesinvarioustissues.[Keywords] Atherosclerosis;Macrophages;Glucocorticoids;Catecholamines;Stress,psychological;Review基金项目:国家重点研发计划项目(2020YFC2003000);国家自然科学基金项目(51672030);中国医学科学院医学科学创新基金项目(2018 I2M 1 002);中央卫生科研计划项目(W2017BJ11)作者简介:王立,硕士研究生,Email:wangli001@foxmail.com通信作者:龚涛,主任医师,博士研究生导师,Email:gb20598@sina.com 动脉粥样硬化是一种慢性、多因素性疾病,该过程主要由血管壁脂质积聚和随后的动脉壁炎症反应构成,最终导致动脉粥样硬化斑块形成和破裂[1 2]。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
Vol. 1(1), pp. 001-007, April, 2013 Available online@http://www.uniquejournals.org/URJMMS ©2013 Unique Research Journals Unique Research Journal of Medicine and Medical Science
Review G protein coupled receptors-systems (GPCRs): Illustrative advances from cannabinoid, chemokine and melanocortins receptors
Abdelaziz Ghanemi1,2, Ling He1* and Ming Yan3 1Department of Pharmacology, China Pharmaceutical University, Nanjing 210009, China.
2Department of Pharmacy, Faculty of Medicine, Mentouri University of Constantine, Constantine 25000, Algeria.
3National Drug Screening Laboratory, China Pharmaceutical University, Nanjing, 210009, China.
*Corresponding author. E-mail: heling92@hotmail.com. Tel: +008613851612452. Fax: +86 2583240236. Accepted 10 April, 2013 The pharmacological targeting of the G protein coupled receptors constitutes an important therapeutic approach among the treatments of the cerebral and the neurological diseases. This importance comes from the fact that G protein coupled receptors are implicated in a variety of physiological functions. Furthermore, studies of the pathological aspects of divers diseases have revealed that G protein coupled receptors play key roles in both the pathogenesis and the prognosis of such diseases. Thus, these receptors became a large area of research in medicine, biology, pharmacology, physiology and even genetics. Herein, we have exemplified how cannabinoid, chemokine and melanocortins receptors’ systems may constitute novel targets in several diseases. The reported findings might also constitute starting points to develop new therapies that could start from the biological properties to reach pharmacological applications. Moreover, these advances may provide further elements to design new research methods and optimize laboratories approaches in both pharmacology and neurosciences.
Keywords: G protein coupled receptor (GPCR), chemokines receptor, cannabinoid receptor, melanocortins receptor, nervous system, potential target.
INTRODUCTION G protein coupled receptors (GPCR) represent the most extensively targeted protein class for marketed drugs (Topiol, 2013) and are one of the largest classes of cell-surface receptors (Santulli et al., 2013). It is estimated that 30 % of current clinical pharmaceutical agents available are ligands directed at GPCRs (Santulli et al., 2013). The GPCRs-related discoveries have a diversity of applications in many medical and even non medical fields. For instance, both neohesperidin dihydrochalcone (NHDC) and cyclamate synergistically potentiated cell response to sucrose (Schiffman et al., 1995, Birch, 1999) which is mediated via GPCRs, therefore, further investigations could allow us to reduce sugar contents in beverage and food and come out with new forms of healthy diet products and provide a starting point to develop more molecules for diabetics and other kind of metabolic disorders as well. The complex coupling and the engaged pathways make targeting GPCRs system more important. Indeed, however, the divers’ novel molecules which target GPCRs systems that provided or may provide new therapeutics remain the main researches topic of the GPCRs system and the recent findings are focusing on the high pharmacological importance of modifying such systems. Noticeable researches and publications illustrate the current status of this area. Indeed, psychiatry is among the important 002 Unique Res. J. Med. Med. Sci. fields that have benefited from the advances of the pharmacological targeting of the GPCRs (Ghanemi), 2013a). Furthermore, laboratories observations about compounds and reagents effects on cells and some GPCRs (Ghanemi), 2013b) gave indications about the potential usage of those chemicals to further develop new leads via the drug screening or via the study of the biological properties. The following examples represent selected advances that show the potential that targeting GPCRs has among the modern pharmacotherapies. By GPCRs systems we refer to the receptors, the enzymatic pathways and the biochemical reactions implicated in the related signaling mechanisms. CANNABINOID RECEPTORS SYSTEM WITH NEUROPROTECTIVE PROPERTIES? Several cognitive functions including learning, memory and neuroprotection, in addition of other neurophysiological aspects have been shown to involve both the GPCR systems of Cannabinoid receptors 1 (CB1R) and 2 (CB2R) (Matsuda et al., 1990, Howlett et al., 2002) and the extracellular signal-regulated kinases 1 and 2( ERK1 and ERK2) (Asimaki and Mangoura, 2011). CB1 receptors exist in different brain regions such as cerebral cortex, striatum, cerebellum, hippocampus and hypothalamus (Herkenham, 1991) and these receptors interact with numerous neurotransmitters (Di Marzo, 2008) thus, it is getting more importance in pharmacology and neurophysiology. Several papers pointed that CB1 receptors activate ERK (Bouaboula et al., 1995, Galve-Roperh et al., 2002, Davis et al., 2003, Derkinderen et al., 2003, Rubovitch et al., 2004). In addition, whereas cell proliferation, transformation, and apoptosis have been linked to CB1R-dependent activation of ERK(Bouaboula et al., 1995, Galve-Roperh et al., 2002), ERK activity has been linked to neuronal differentiation, synaptic plasticity and memory (Kelleher et al., 2004, Samuels et al., 2008). A recent study, that aimed to indentify the enzymatic pathway events that link CB1R to the activation of ERK in CNS neurons (Asimaki and Mangoura, 2011), has highlighted that both PKC (precisely PKCε) and Tyrosine kinases TK (including the two families tyrosine-protein kinase Src (Src), and tyrosine-protein kinase Fyn (Fyn)) are implicated in the CB1R pathway. These results are consistent with previous data. Indeed, PKCε has many roles in CNS including learning and memory and in synaptic remodeling (Hama et al., 2004). It has also been found prominently in differentiating and differentiated neurons (Prekeris et al., 1996, Mangoura et al., 2006) supposing a role in neuron differentiation. Furthermore, numerous studies have linked CB1Rs with PKC activity (Hillard and Auchampach, 1994, Rubovitch et al., 2004, Wallace et al., 2009). On the other hand, other publications have indicated that GPCR signaling may involve tyrosine kinases(Ma et al., 2000, Amorino et al., 2007) (Mangoura and Dawson, 1993, Rodriguez-Fernandez and Rozengurt, 1996, Mangoura, 1997, Sandilands et al., 2007) and the same enzyme(PKC) was precisely shown to play a role in CB1R activity (Galve-Roperh et al., 2002; Derkinderen et al., 2003; Hart et al., 2004; Rubovitch et al., 2004; He et al., 2005). Recently, the CB₂ cannabinoid receptor ligands 4-Oxo-1,4-dihydropyridines have shown a protection against experimental colitis (El Bakali et al., 2012) and both albuminuria and podocyte protein loss can be ameliorates by CB2 activation (Barutta et al., 2011). Furthermore, CB2R Agonist may ameliorate Alzheimer-Like Phenotype (Aso et al., 2013) . Taken together, these new elements illustrate the neuroprotective properties of CB1R and the enzymes implicated in its pathway (ERK, PKC and TK), that are the elements via which the signal is transferred from the GPCR to the endocellular medium, and how they are linked with several neurofunctions such us learning and memory; in addition of some neurophysiological processes including cell proliferation, transformation and synaptic plasticity. Therefore, targeting CB1R or the related enzymatic arsenal may provide new therapeutic agents for CNS-related diseases, mainly those with neurodegenerative components and those with the loose of cognitive functions as symptoms. Although, herein we provide a starting point yet deeper researches and investigations are still required.