微丝细胞骨架重塑异常与糖毒性导致的胰岛β细胞分泌 功能障碍
毒作用机制

2. 亲核外源化学物在过氧化物酶作用下丢失一 个电子,后者可形成含氧自由基
如:氢醌(亲核外源化学物)连续发生两次单电子转移产 生半醌自由基与含氧自由基。
3. 电子向分子转移引起的还原性键均裂
如:四氯化碳从细胞色素P450或线粒体电子传递链获得一 个电子,还原脱氯,生成CCI3. ;H2O2均裂产生HO· 。
蛋白质、核酸、脂质等生物大分子。
羟基 巯基 ε-氨基 胍基 咪唑基等
碱基 核糖 脱氧核糖 磷酸脂
3.去氢反应
如内源性巯基化合物(R-SH)在自由基作用下去氢形
成巯基自由基(R-SH.) 。
4.电子转移
外源化学物使Hb分子中的Fe2+氧化成Fe3+ ,引起高铁血 红蛋白症,如亚硝酸盐。
5.破坏酶促反应
(四)内质网应激(endoplasmic reticulum stress)
内质网是细胞蛋白质和脂质合成、加工、折叠和运输的 重要场所,内质网在外来因素刺激下发生应激反应。
特异性分子伴侣:糖调节蛋白78
(glucose regulated protein 78, GRP78) 介导内质网应激的跨膜蛋白(三个): 肌醇需求酶1(inositol-reguiring enzyme1,IRE1) 蛋白激酶R样内质网激酶 (protein kinase R-like ER kinase, PERK) 激活转录因子6 (the activating transcription factor 6, ATF6)
一、反应类型
1.非共价结合(nonconvalent binding) 终毒物通过非极性交互作用或以氢键与离子 键等非共价结合方式与内源性分子结合。 被结合的内源性分子有:
认知障碍的病因及发病机制

认知障碍的病因及发病机制认知是大脑皮层复杂高级功能的反映,任何直接或间接导致大脑皮层结构和功能慢性损伤的因素均可通过不同机制引起认知障碍,现将其归纳如下:(一)慢性脑损伤1.脑组织调节分子异常(1)神经递质及其受体异常:大多数神经元之间的信息传递是通过神经递质(neurotransmitter)及其相应的受体完成的。
这些神经递质或受体异常改变均可导致不同类型和不同程度的认知异常。
1)多巴胺(dopamine):多巴胺是以酪氨酸为底物,在酪氨酸羟化酶(tyrosine hydroxylase)和多巴脱羧酶(dopamine decarboxylase)的作用下合成的。
研究发现:脑中多巴胺含量显著降低时可导致动物智能减退、行为情感异常、言语错乱等高级神经活动障碍。
例如,在帕金森病(Parkinson disease,PD)患者黑质多巴胺能神经元减少,酪氨酸羟化酶和多巴脱羧酶活性及纹状体多巴胺递质含量明显卞降。
此外,在动物实验中发现多巴胺过多也可导致动物认知功能的异常改变。
多巴胺受体有D1和D2受体两大家族,精神分裂症患者与大脑额叶皮层的D1受体功能低下和皮层下结构D2受体功能亢进双重因素有关,因此有人提出用D1激动和D2阻断治疗精神分裂症的新概念。
2)去甲肾上腺素(nonepinephrine):去甲肾上腺素是最早被发现的单胺类神经递质,是多巴胺经β羟化酶作用生成的产物。
在脑,去甲肾上腺素通过α1、α2和β受体发挥调节作用。
在突触前,α2受体通过Gi蛋白介导,减少cAMP的生成和cAMP依赖性蛋白激酶的活性,减少蛋白激酶对N-型Ca2+通道的磷酸化,以至Ca2+通道关闭,Ca2+流减少,从而对去甲肾上腺素的释放起抑制作甩(负反馈调节);α2受体激动还可抑制在警醒状态下的蓝斑神经元的放电增加;在突触后,α1受体激动可引起K+通道开放,K+外流增加,神经元倾向超极化而产生抑制效应。
而α1受体激活则使K+通道功能降低,K+外流减少,神经元去极化产生兴奋效应。
胰岛素药物的临床稳定性的影响因素及其对策分析

胰岛素药物的临床稳定性的影响因素及其对策分析摘要:胰岛素是含有蛋白质和肽类激素、开启后可多次使用的一种多剂量注射药物,是治疗糖尿病的主要药物,既可调节血糖又能促进机体代谢。
胰岛素的药物稳定性是决定糖尿病患者治疗效果的关键因素,包括物理稳定性(如温湿度等影响胰岛素的活性)、化学稳定性(如氧化水解等反应导致胰岛素化学降解)和生物稳定性(如微生物的滋长加速胰岛素腐败),任何一种稳定性被破坏均会造成胰岛素的效价降低。
目前我国仍有部分研究对胰岛素的药物稳定性认识不足,保存、使用和管理不规范,胰岛素开启后的储存时间备受争议,过去的研究主要集中于所有药物制剂稳定性,忽略了胰岛素这类药物的特殊性。
因此,本文将全面分析胰岛素药物稳定性的影响因素及胰岛素的干预性策略,旨在提升临床使用胰岛素制剂的稳定性、安全性和药物管理的规范性,为确定胰岛素开启后的有效期提供参考,确保糖尿病患者治疗效果。
关键词:胰岛素;药物稳定性;影响因素引言糖尿病主要由胰岛素抵抗和胰岛β细胞功能降低引起,是一种常见的内分泌代谢疾病,具有遗传易感性,容易在环境因素的触发下发病。
胰岛素既可以提供降低餐后血糖的速效胰岛素成分,又可以提供降低空腹血糖和基础血糖的中效胰岛素成分。
研究观察胰岛素在临床治疗糖尿病的安全性和疗效,证实血糖控制不佳的中国糖尿病患者,无论是否已经接受过口服降糖药治疗,接受胰岛素治疗24周后耐受性和安全性均良好,同时显著改善了血糖控制。
另外一项国际开放、非随机、非干预的临床观察性试验研究同样表明,胰岛素起始治疗26周后,患者的HbA1c、空腹血糖水平和重度低血糖事件发生概率均显著下降,从而也证明了胰岛素在糖尿病患者起始治疗中可以兼顾有效性和安全性。
目前大量研究表明,胰岛素在临床治疗糖尿病具有良好的安全性、方便性和有效性,值得临床推广应用。
所以,胰岛素在临床使用过程中稳定性的研究具有非常重要的意义。
1胰岛素治疗T2DM患者可明显改善患者的血糖水平,减小患者血糖波动幅度,缩短血糖达标时间及减少达标时使用胰岛素总剂量,降低治疗过程中低血糖发生率。
细胞骨架和内吞作用的抑制、激活剂
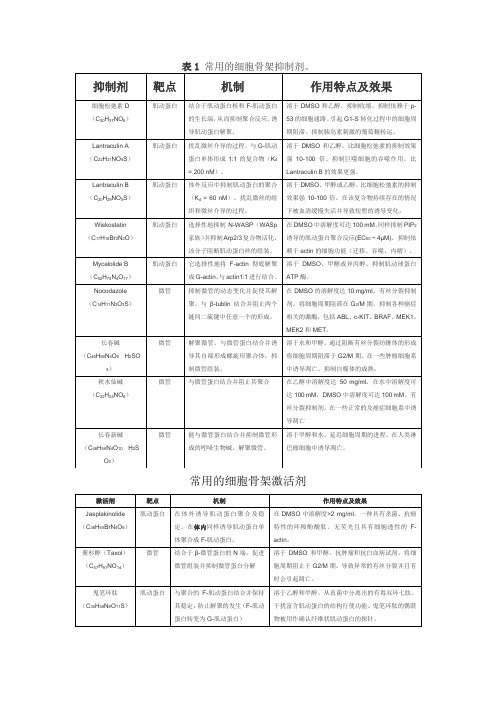
溶于DMSO和乙醇。同样抑制Drp1(线粒体)。会引起纤维化并在胸膜间皮细胞中诱导PAI-1。抑制BSC1细胞的细胞铺展和迁移。
非律平(C35H58O11)
胞膜窖介导的内吞作用
与膜上的胆固醇结合并形成超微机构聚集与复合。
非律平由4个异构的多烯大环内酯组成。非律平III是主要成分。抗生素和抗真菌。抑制朊蛋白(PrP)的内吞。
鬼笔环肽(C35H48N8O11S)
肌动蛋白
与聚合的F-肌动蛋白结合并保持其稳定,防止解聚的发生(F-肌动蛋白转变为G-肌动蛋白)
溶于乙醇和甲醇。从真菌中分离出的有毒双环七肽。干扰富含肌动蛋白的结构行使功能。鬼笔环肽的偶联物被用作确认纤维状肌动蛋白的探针。
表常用的内吞作用抑制剂。HOE.64和LY294002是另外两种巨胞饮抑制剂。单丹磺酰尸胺和巴弗洛霉素A1是用于研究网格蛋白介导的内吞作用的另外两种抑制剂。
在DMSO中溶解度可达100 mM。同样抑制PIP2诱导的肌动蛋白聚合反应(EC50~ 4μM)。抑制依赖于actin的细胞功能(迁移、吞噬、内褶)。
Mycalolide B(C52H74N4O17)
肌动蛋白
它选择性地将F-actin彻底解聚成G-actin。与actin1:1进行结合。
溶于DMSO、甲醇或异丙醇。抑制肌动球蛋白ATP酶。
肌动蛋白
体外反应中抑制肌动蛋白的聚合(Kd= 60 nM)。扰乱微丝的组织和微丝介导的过程。
溶于DMSO、甲醇或乙醇。比细胞松弛素的抑制效果强10-100倍。在该复合物持续存在的情况下被血清缓慢失活并导致短暂的诱导变化。
Wiskostatin(C17H18Br2N2O)
肌动蛋白
第4章 毒作用机制(150915)

11
二、 从血液循环进入靶部位
※ (二)妨碍毒物分布到靶部位的机制
1. 血浆蛋白结合:与高分子血浆蛋白或脂蛋白结合,影响 扩散。
2. 专一化屏障:血脑屏障、血睾屏障、胎盘屏障阻止亲水 性化合物 3. 贮存部位分布:铅取代羟磷灰石中的钙离子沉积在骨骼、 氯代烃蓄积脂肪细胞 4. 与细胞内结合蛋白结合:金属硫蛋白与镉结合 5. 从细胞内排出:mdr编码的P蛋白将化合物泵回细胞间隙
很容易与谷脱甘肽反应而解毒
30
(二)解毒
4. 自由基的解毒 没有任何一种可以解除HO〃,预防HO〃毒作用的 最有效办法是阻止其产生。
超氧化物歧化酶(SOD),谷胱甘肽氧化酶(GPO)和过氧化氢酶 (CAT)对超氧阴离子自由基(O2〃-)的解毒作用
31
(二)解毒 5. 蛋白质毒素的解毒 硫氧环蛋白,蛇毒 6. 解毒过程失效 解毒能力耗竭(解毒酶、共底物、抗氧化剂);
3
阐明毒作用机制的意义:
(1)解释描述性资料、评估特定外源化学物引起有害效
应的概率、制定预防策略、设计危害程度小的药物和工业 化学物,以及开发靶生物具有良好选择性的杀虫剂等提供 依据; (2)有利于对机体生理和生化过程及人类某些疾病重要 疾病病理过程的进一步认识。
4
毒物在体内的可能毒性过程
生的防御或适应性反应。 根据引起细胞应激的原因不同以及细胞应激反应的差异: • 热应激(heat stress); • 氧化应激(oxidative stress);
• 缺氧应激(hypoxic stress);
• 内质网应激(endophasmic reticulum stress); • 遗传毒性应激(genotoxic stress)
游离脂肪酸对胰岛素分泌的影响

游离脂肪酸对胰岛素分泌的影响班级:02级口腔姓名:任抒欣指导教师:王子梅徐锦【摘 要】循环游离脂肪酸(FFA) 水平升高以及TG在β细胞内的堆积,生理范围内可通过改变Ca2+浓度、胰岛素分泌信号传导通路等机制调节基础胰岛素分泌增加,游离脂肪酸浓度长时间超过生理范围时,β细胞内胰岛素含量逐渐下降,并出现β细胞凋亡增加,细胞数量减少。
神经酰胺依赖及非依赖的途径均参与了脂性凋亡的过程。
【关键词】游离脂肪酸;胰岛素;凋亡【Abstract】The elevations of plasma FFA levels and the accumulation of TG In pancreatic β-cells of physical level potentiate glucose-stimulated insulin secretion. The long-time accumulation of FFA levels decreases insulin secretion and stimulates β-cells apoptosis. The metabolism of this process will be discussed in the article.【Key words】free fatty acid; insulin; apoptosis近年来,在II型糖尿病的发病机制中提出了脂毒性的概念,由于脂毒性与葡萄糖毒性有许多交叉,因此称之为“糖脂毒性(glucolipotoxicity)”或“脂糖毒性(lipoglucotoxicity)”。
“脂毒性”是指血循环中游离脂肪酸浓度过高以及细胞内脂质含量过多(主要是肌细胞、肝细胞、胰岛β细胞)所引起的致糖尿病作用。
“脂毒性”的致糖尿病作用既可以引起β细胞分泌胰岛素功能缺陷,又导致胰岛素抵抗的发生。
在生理状态下,游离脂肪酸对β细胞分泌胰岛素起促进作用,但实验证明,长期暴露于高浓度的游离脂肪酸中,β细胞易发生功能损伤和凋亡。
水飞蓟素治疗脂肪肝的作用机制
水飞蓟素治疗脂肪肝的作用机制【关键词】脂肪肝系脂肪肝系指各种原因所致的肝脏脂肪(主要为三酰甘油)蓄积过多、代谢平衡失调的病理态。
临床上常根据饮酒与否,将其分为酒精性脂肪肝和非酒精性脂肪肝。
尽管病因不同,二者却具有相似的病理学变化。
目前对脂肪肝的治疗尚缺乏针对性的特效药,主要是祛除原发病因及戒酒、减肥、饮食运动等综合治疗,适当应用降脂保肝药物治疗。
水飞蓟素(silymarin)是菊科草本植物水飞蓟种子中提取的类黄酮的有效活性成分,据现代药理研究水飞蓟素可直接清除自由基、对抗脂质过氧化、促进被损伤肝细胞合成DNA及结构蛋白、免疫调节和抗肝纤维化等药理活性,是经典的肝损伤修复药物,在当前尚缺乏其他有效的治疗脂肪肝药物的情况下,它在这方面的作用令人瞩目。
本文就此进行综述。
1 脂肪肝的形成1.1 酒精性脂肪肝饮酒后,乙醇在胃肠很快被吸收,肝脏是乙醇代谢的唯一器官。
90%的乙醇经过肝脏作用变为乙醛,乙醛再变成乙酸。
酒精性脂肪肝发生的机制可能有以下几个因素的参与[1]。
1.1.1 酒精对肝损害导致脂肪肝的主要原因酒精是通过ADH和ALDH氧化,在此过程中产生大量的尼克酰胺腺嘌呤二核苷酸脱氢酶(NADH),使NADH与尼克酰胺腺嘌呤二核苷酸(NAD)比值高。
这就抑制了线粒体三羧酸循环,使脂肪酸代谢发生障碍,氧化减弱;同时促进脂肪酸合成,从而使脂肪在肝细胞内堆积而发生脂肪变性,最终导致脂肪肝形成,这是酒精对肝损害导致脂肪肝的主要原因。
1.1.2 酒精所致的肝损害中P450 2E1诱导和激活的巨噬细胞是氧自由基的重要来源由于乙醛的毒性大于乙醇,而前者可促进脂质过氧化,产生线粒体损害,使还原型谷胱甘肽(GSH)减少,并增加胶原合成。
乙醛和另一些脂质过氧化产物如丙二醛(MDA)等能形成蛋白加合物,引起免疫反应,损害细胞骨架功能和细胞与间质的相互作用,从而抑制重要的代谢通路。
在慢性酗酒者中,细胞色素P450 2E1能高度诱导ADH。
阿尔兹海默症动物模型概述
动物模型
01
以衰老为基础 的AD模型
自然衰老的动物模型 快速老化小鼠模型
02
各种因素诱发 的AD模型
化学损伤
物理损伤
饮食诱导
03
转基因AD模型
APP 转基因模型 PS1 转基因模型 tau 相关模型 多重转基因模型
以衰老为基础的AD模型
AD 多发于老年人,衰老型 AD 动物模型是根据年龄老化为 AD 的重要危险因素之一而建立,主要 为自然衰老、快速性衰老模型 。
缺点:可以模拟AD的临床症状,但缺乏AD特异性胆碱神经损伤以及Aβ 沉积。且由于创伤较大,不相关的干扰因素过多,易引起脑内其他部位的 损伤及造模动物的死亡。因此,该模型成功率低,现在已很少使用。
饮食诱导AD 模型
高脂饮食诱导模型
有报道指出动物给予高脂饲料饲养可降低大脑对葡萄糖的摄取,诱导动物模型产生糖耐量降低及胰岛素抵 抗,亦可损伤神经元胰岛素受体功能,引起tau 蛋白过度磷酸化,从而导致 NFT。
神经细胞- AD 谷氨酸
Bcl-2 Bax
caspase
神经细胞凋亡
Ca+内流-----细胞内钙离子超载 -----细胞超微结构破坏 膜电位改变
以化学为基础的AD模型
秋水仙碱诱导模型
秋水仙碱可选择性破坏海马神经元,降低乙酰胆碱酯酶 (AChE)活性,破坏胆碱能神经通路,使动物出现短期学 习记忆障碍。秋水仙碱诱导的AD大鼠海马中发现了染色体 溶解和淀粉样蛋白斑块以及高水平的 ROS、亚硝酸盐、 TNF-α.
蛋白质运输与细胞骨架
蛋白质运输:将信号 分子从细胞膜传递到
细胞核或细胞质
信号转导:细胞对外界 刺激的反应和传递
细胞骨架:维持细胞形 态,参与细胞运动和分
裂
相互作用:蛋白质运 输和细胞骨架共同参 与信号转导,调控细
胞功能
蛋白质运输与疾病治疗的关系
蛋白质运输异常与疾病的发生和 发展
蛋白质运输在疾病治疗中的应用
蛋白质运输与疾病治疗的未来研 究方向
蛋白质运输与细胞骨架的研究将继续深入,可能会发现更多新的蛋白质和细 胞骨架成分。 蛋白质运输与细胞骨架的研究可能会为治疗某些疾病提供新的思路和方法。
蛋白质运输与细胞骨架的研究可能会为药物研发提供新的靶点。
蛋白质运输与细胞骨架的研究可能会为细胞生物学领域的发展提供新的动力。
汇报人:XXX
下一次运输
蛋白质运输异常可能导致疾病 蛋白质运输与神经退行性疾病的关系 蛋白质运输与癌症的关系 蛋白质运输与免疫疾病的关系
微管:主要成分是蛋 白质,负责细胞内的 物质运输和细胞分裂
中间纤维:主要成分是 蛋白质,负责细胞的形
状和运动
微丝:主要成分是蛋白 质,负责细胞的运动和
分裂
核骨架:主要成分是 蛋白质,负责细胞的 核内物质运输和核分
蛋白质运输与细胞骨架的相互作用 影响细胞添加标题
添加标题
蛋白质运输影响细胞骨架的组装和 拆卸
蛋白质运输异常可能导致细胞骨架 病变,影响细胞功能
蛋白质运输:在细 胞分裂过程中,蛋 白质需要被运输到 正确的位置,以参 与细胞分裂的各个 步骤。
细胞骨架:细胞骨 架在细胞分裂过程 中起着重要的作用, 它帮助细胞保持形 状,参与细胞分裂 的各个步骤。
细胞骨架在药物 研发中的重要性
细胞骨架与药物 靶点的关系
2_型糖尿病患者血清肿瘤标志物CA19-9、CEA_水平与糖化血红蛋白、血糖、C_肽、脂类等指标的相
DOI:10.16658/ki.1672-4062.2023.04.0172型糖尿病患者血清肿瘤标志物CA19-9、CEA 水平与糖化血红蛋白、血糖、C肽、脂类等指标的相关性研究梁碧1,陈月云2,蔡月娣1,覃东红1,缪媛媛11.广东省农垦中心医院内分泌科,广东湛江524000;2.湛江麻章辉文医院内科,广东湛江524000[摘要]目的2型糖尿病患者血清肿瘤标志物CA19-9、CEA水平与糖化血红蛋白、血糖、C肽、脂类等指标的相关性探讨。
方法将2020年7月—2022年8月广东省农垦中心医院接诊的300例2型糖尿病患者纳入研究组,选取同期参与健康体检的300名健康人纳入对照组,分析血清肿瘤标志物CA19-9、CEA水平与糖化血红蛋白、血糖、C肽、脂类等指标的相关性。
结果研究组患者的CA19-9、CEA、HbA1c、FPG、TG指标高于对照组,HDL指标低于对照组,差异有统计学意义(P<0.05)。
两组TC与LDL指标对比,差异无统计学意义(P> 0.05)。
血清肿瘤标志物与糖脂代谢指标相关性分析显示,HbA1c、FPG、2 hPG与CA19-9及CEA呈正相关(P< 0.05),C0、C2与CEA呈负相关(P<0.05)。
结论2型糖尿病患者血清肿瘤标志物CA19-9、CEA水平比健康者更高,且与HbA1c、血糖及C肽有一定相关性。
[关键词] 2型糖尿病;血清肿瘤标志物;CA19-9;CEA;糖化血红蛋白;血糖[中图分类号] R587.1 [文献标识码] A [文章编号] 1672-4062(2023)02(b)-0017-04Study on the Correlation between Serum Tumor Markers CA19-9, CEA and Glycosylated Hemoglobin, Blood Glucose, C-peptide, Lipids and Other Indicators in Patients with Type 2 DiabetesLIANG Bi1, CHEN Yueyun2, CAI Yuedi1, QIN Donghong1, MIAO Yuanyuan11.Department of Endocrinology, Agricultural Reclamation Central Hospital, Zhanjiang, Guangdong Province, 524000 China;2.Internal Medicine, Zhanjiang Mazhang Huiwen Hospital, Zhanjiang, Guangdong Province, 524000 China[Abstract] Objective To explore the correlation between serum tumor markers CA19-9, CEA and glycosylated hemo⁃globin, blood glucose, C-peptide, lipids and other indicators in patients with type 2 diabetes. Methods 300 patients with type 2 diabetes who were admitted to Guangdong Provincial Agricultural Reclamation Center Hospital from July 2020 to August 2022 were included in the study group, and 300 healthy people who participated in health examina⁃tion at the same time were selected as the control group. To analyze the correlation between serum tumor markers CA19-9 and CEA levels and indicators such as glycosylated hemoglobin, blood sugar, C-peptide, and lipids. Results The CA19-9, CEA, HbA1c, FPG, and TG indexes of patients in the study group were higher than those in the control group, while the HDL indexes were lower than those in the control group, and the difference was statistically signifi⁃cant (P<0.05) There was no statistically significant difference in TC and LDL between the two groups (P>0.05). The correlation analysis between serum tumor markers and glycolipid metabolism indicators showed that HbA1c, FPG, and 2 hPG were positively correlated with CA19-9 and CEA (P<0.05), C0 and C2 were negatively correlated with CEA (P< 0.05). Conclusion The levels of serum tumor markers CA19-9 and CEA in patients with type 2 diabetes were higher than those in healthy people, and had some correlation with HbA1c, blood glucose and C-peptide.[基金项目]2021年湛江市科技计划项目(2021B01256)。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
·专题笔谈·微丝细胞骨架重塑异常与糖毒性导致的胰岛β细胞分泌功能障碍严丹马晓松糖毒性(glucotoxicity)的重要特征之一是葡萄糖刺激的胰岛素分泌消失,然而其机制迄今仍不清楚。
大量的实验证据表明,β细胞内微丝(F-actin)的解聚是胰岛素分泌的前提。
微丝是细胞骨架成分之一,在囊泡的胞内转运和出胞、细胞形态的维持、细胞迁移等过程中起重要作用。
在生理条件下,微丝在β细胞内形成致密的网状结构,阻碍胰岛素囊泡的锚泊(docking)和出胞(exocytosis)。
葡萄糖刺激可使微丝解聚,诱导胰岛素分泌。
我们近期的工作表明慢性高糖培养导致微丝细胞骨架聚集,且抵抗葡萄糖诱导的微丝骨架的解聚,提示微丝细胞骨架重塑异常可能是糖毒性损害胰岛β细胞分泌功能的机制之一。
一、葡萄糖刺激胰岛素囊泡出胞的机制胰岛素在胰岛β细胞内合成并储存于分泌囊泡中,胰岛素囊泡存在于两类不同的功能池:易释放池(readily releasable pool)和储存池(reservepool)[1]。
大约1%~5%的分泌囊泡存在于易释放池,该池的囊泡经过获能(priming)已具备释放能力,且锚泊于细胞膜。
升高胞浆Ca2+浓度可触发易释放池囊泡快速出胞。
其余95%~99%的胰岛素囊泡位于距释放部位较远的储存池。
储存池囊泡不具备释放能力,它们必须经过获能取得释放能力,并经胞内运输(translocation)转运至易释放池才能被释放。
胰岛素囊泡通过Ca2+-触发的出胞将胰岛素分泌出胞外。
胰岛素囊泡的出胞是由囊泡膜与细胞膜的融合(fusion)所致,这一过程由可溶性N-乙基马来酰亚胺敏感因子结合蛋白受体(SNAREs)介导。
SNAREs家族蛋白包括两组成员:位于细胞膜的靶蛋白(t-SNAREs:syntaxin和SNAP-25)和位于囊泡膜的囊泡蛋白(v-SNARE:VAMP2)[2]。
胞浆Ca2+浓度升高触发VAMP-2与syntaxin/SNAP-25形成核心复合物(core complexes),通过“拉链”机制使囊泡膜与其对应的细胞膜融合,导致胰岛素的释放[1]。
SNARE蛋白不仅介导囊泡的出胞过程,而且他们与Ca2+通道直接连接而影响Ca2+内流。
研究发现,连接L-型Ca2+通道第2和第3同源域的内环(L-loop)与syntaxin、SNAP-25及VAMP-2相连,使Ca2+通道与易释放池胰岛素囊泡构成Ca2+通道-囊泡“释放复合体”[3]。
正是由于以上结构基础,当Ca2+通道开放时,锚泊于细胞膜的易释放池囊泡迅速暴露于紧邻Ca2+通道入口的高Ca2+环境,触发膜融合机制引发囊泡出胞。
在生理条件下葡萄糖是调控胰岛素囊泡出胞的最重要因素。
低血糖(5mmol/L)时,β细胞代谢水平低,胞浆低浓度的ATP导致胞膜K ATP通道开放,使β细胞处于静息状态;血糖水平升高(≥7 mmol/L),β细胞通过胞膜上的葡萄糖载体-2(GLUT-2)将葡萄糖摄入胞内,葡萄糖在线粒体代谢升高胞浆ATP/ADP的比率,关闭胞膜K ATP通道,使细胞膜去极化,继而开放电压门控Ca2+通道,Ca2+内流触发胰岛素囊泡的出胞[1]。
二、β细胞内微丝在胰岛素分泌中的作用微丝,亦称纤维型肌动蛋白,是由球型肌动蛋白(G-actin)单体通过成核和延长等过程形成的纤维型多聚体。
β细胞超微结构的结果显示,微丝在细胞膜下形成网状结构,从而阻碍胰岛素囊泡与细胞膜的接触,抑制胰岛素的分泌。
近年来的研究发现,在β细胞内微丝与t-SNARE家族的SNAP-25、syntaxin1、syntaxin4相结合(与syntaxin4是直接结合)[4],进而干扰囊泡膜上的v-SNARE与细胞膜上的t-SNARE结合,阻碍囊泡的锚泊、获能和与细胞膜的融合。
微丝与SNARE结合的重要特征是:球形肌动蛋白单体不具备与t-SNARE结合的能力,只有聚集成纤维型多聚体才能与SNARE结合。
因此,解聚微丝可导致其与SNAP-25和syntaxin的解离,从而增强葡萄糖刺激的胰岛素分泌[5]。
相反,促进微丝聚集或稳固微丝多聚体结构(用微丝稳定因子jasplakinolide处理)可抑制葡萄糖诱导的微丝解聚,同时也抑制其刺激胰岛素分泌的作用[5]。
由此DOI:10.3760/cma.j.issn.1674-5809.2014.08.000作者单位:518060深圳大学糖尿病研究所通信作者:马晓松,Email:xsma@可见,微丝细胞骨架的重塑是影响胰岛素囊泡出胞的重要环节。
在生理条件下,葡萄糖刺激导致微丝解聚并使其与t-SNARE解离,进而诱导胰岛素囊泡的出胞。
葡萄糖解聚微丝的具体机制目前仍不十分清楚。
研究表明,葡萄糖诱导微丝细胞骨架重塑与其激活细胞外信号调节激酶(ERK1/2)[6]和细胞骨架蛋白黏着斑激酶(FAK)[7]密切相关。
此外,ERK1/2的激活导致微丝解聚;FAK的激活导致微丝与t-SNARE 解离,使t-SNARE与囊泡的v-SNARE结合,有助于囊泡锚泊与出胞。
三、糖毒性与胰岛β细胞分泌功能障碍2型糖尿病慢性持久的高血糖直接损害胰岛β细胞功能,这一现象称为糖毒性。
糖毒性所致胰岛素的分泌降低又进一步升高血糖,使糖毒性加剧,从而加速糖尿病进程并导致糖尿病并发症的发生。
临床资料显示2型糖尿病患者经治疗血糖恢复正常后,β细胞功能也随之恢复。
动物实验研究发现:降低葡萄糖水平可纠正糖尿病大鼠的β细胞功能异常,并使胰岛素分泌恢复正常。
离体高糖环境培养β细胞系INS-1细胞5d(20mmol/L葡萄糖)或2d(30mmol/L葡萄糖),均导致葡萄糖刺激的胰岛素分泌明显减弱,说明慢性持久的高糖直接抑制胰岛β细胞的分泌能力;降低葡萄糖水平可恢复β细胞功能并纠正胰岛素分泌紊乱,提示糖毒性持久慢性高糖直接抑制了胰岛β细胞的分泌功能。
Dubois等[8]对糖毒性条件下INS-1E细胞内后高尔基体囊泡(post-Golgi vesicles)转运/出胞相关的30多个蛋白进行表达量检测,发现一些与Ca2+诱导囊泡出胞的相关蛋白下调,例如VAMP-2、syntaxin1和complexin,并且发现VAMP-2在人胰岛中表达量也下降。
相比之下,其他与后高尔基体囊泡转运对应的成分被保留且细胞未发生脱颗粒(degranulated),证实糖毒性的确干扰了胰岛素囊泡的出胞过程。
四、糖毒性增加β细胞内的应力纤维应力纤维(stress fibre)是由10~30根微丝交联形成的束状结构,其末端连接在整合素富集的细胞膜黏着斑,因此应力纤维长且结构较稳固。
应力纤维的形成主要由小分子三磷酸鸟苷(GTP)酶Rho 家族蛋白调控。
Rho家族中的3个成员-RhoA、RhoB和RhoC中,Rho A是应力纤维形成的主要调控因子[9]。
Rho A通过激活其下游效应分子Rho-相关蛋白激酶(ROCK)和成核蛋白Formin1促进微丝聚集,形成应力纤维[10]。
ROCK是丝氨酸/苏氨酸激酶,其被激活后可直接磷酸化肌球蛋白轻链2(myosin light chainⅡ,MLC2)19个丝氨酸位点,此位点被ROCK磷酸化后可以增加肌球蛋白Ⅱ(myosinⅡ)ATPase活性从而增加应力纤维的收缩性。
在另一方面,ROCK可磷酸化MBS(MYPT)-肌球蛋白轻链磷酸酶(myosin light chain phosphatase)调节亚基,ROCK诱导的MBS磷酸化可抑制肌球蛋白轻链磷酸酶(MLCP)的活性而使MLC2磷酸化增加和应力纤维形成,Formin1则使平行排列的微丝聚集。
此外,ROCK通过激活LIM域激酶1抑制微丝解聚因子(cofilin、ADF)[10],阻止微丝解聚,也有助于应力纤维的形成。
Tomas等[11]在激光共聚焦显微镜下观察到对葡萄糖刺激敏感β细胞(MIN6细胞系的B1细胞)内微丝细胞骨架以松散的肌动蛋白纤维网状结构为主,葡萄糖刺激可导致纤维网状结构解聚,诱导胰岛素分泌。
而另一类对葡萄糖刺激无反应的β细胞(MIN6细胞系的C3细胞),微丝细胞骨架则以长且粗大的应力纤维为主;葡萄糖刺激不能解聚应力纤维,也不能诱导胰岛素分泌。
与C3细胞类似,我们近期发现慢性高糖培养(含30.0mmol/L葡萄糖的培养基培养72h),β细胞(包括INS-1细胞、MIN6细胞和小鼠β细胞)内微丝聚集形成大量应力纤维;而正常培养的β细胞(含5.5mmol/L葡萄糖的培养基培养72h)微丝细胞骨架以短、细且松散的纤维丝为主,应力纤维很少。
特别值得注意的是,以应力纤维为主的胰岛β细胞对葡萄糖刺激不敏感:葡萄糖刺激不能诱导应力纤维的解聚,也不能刺激胰岛素分泌。
用微丝解聚剂lutrunculin B解聚应力纤维,可恢复葡萄糖刺激的胰岛素分泌。
由此可见,糖毒性β细胞增多的应力纤维与葡萄糖刺激胰岛素分泌的消失密切相关。
不难理解微丝解决可促使囊泡v-SNARE与细胞膜t-SNARE结合,导致囊泡锚泊与获能,在Ca2+触发下导致囊泡膜与细胞膜融合,胰岛素的释放。
五、胰高血糖素样肽-1(GLP-1)改善糖毒性β细胞的功能GLP-1由肠道L-细胞分泌,其作用的主要靶点之一是胰岛β细胞。
生理条件下,GLP-1通过增加胰岛素含量,促进β细胞的增殖及抑制其凋亡来增强胰岛β细胞的功能。
此外,GLP-1最显著的生物学作用是增强胰岛β细胞的分泌能力。
在正常生理条件下,GLP-1增强β细胞胰岛素分泌是由环磷酸腺苷(cAMP)-被腺苷酸激活的交换蛋白2(exchange protein activated by cAMP,Epac2)和cAMP-蛋白激酶A(PKA)这两条通路所介导[12-13]。
GLP-1受体(GLP-1R)是兴奋型G蛋白(Gs)耦联受体,GLP-1通过与其受体结合继而激活腺苷酸环化酶,升高胞内cAMP[12],进而激活Epac2和PKA增进胰岛β细胞分泌功能。
cAMP-Epac2和cAMP-PKA 介导GLP-1增强分泌的作用主要通过升高胞浆内Ca2+浓度、加速胰岛素囊泡从储存池向易释放池的转运及胰岛素囊泡出胞过程实现的。
文献报道,GLP-1增强胰岛素分泌的效能中50%~60%由cAMP-PKA信号通路介导,40%~50%则由cAMP-Epac2通路介导[13],同时抑制Epac2和PKA则完全阻断GLP-1或forskolin(AC激动剂)增强的胰岛素分泌。
然而在糖毒性条件下,GLP-1增进胰岛素分泌机制则鲜有报道。
我们之前的工作[14]揭示GLP-1恢复糖毒性β细胞葡萄糖刺激的胰岛素分泌,其效应继发于提高胞浆cAMP浓度并主要由cAMP-PKA 信号通路介导。
近来我们进一步发现GLP-1可解聚糖毒性β细胞应力纤维并恢复糖毒性β细胞葡萄糖刺激的胰岛素分泌,而这种效应可以被PKA的激动剂6-BNZ所模仿,因此推测GLP-1改善糖毒性β细胞功能是通过激活cAMP-PKA通路进而解聚糖毒性β细胞应力纤维实现的。