除菌过滤工艺验证
新版GMP对除菌过滤工艺及验证的要求

分类
测试名称
测试实施者
破坏性
细菌挑战测试
起泡点测试,扩散测试 ,保压实验,水侵入法
制造商
非破坏性
制造商及使用者
现代中药制药论坛搜集
应该在什么时候做完整性测试?
n
灭菌前?
n
灭菌后,过滤前使用前?
n
过滤后?
什么时候检测完整性?
现代中药制药论坛搜集
预过滤 澄清过滤
培养基预过 滤及除菌过 滤
除菌过滤
大/小容量注射剂 LVP and SVP
现代中药制药论坛搜集
预过滤
除菌过滤
API原料药
现代中药制药论坛搜集
N FF - Sterilization
气体除菌
澄 N FF - B ulk 清过滤
5
根据过滤阶段
现代中药制药论坛搜集
孔径逐渐减小的过滤系 列
澄清
在过滤系列开始是最大 的过滤容量
预滤
在过滤系列终端是最大 的过滤截留率
除菌
3 根据过滤阶段 Filtration stage
现代中药制药论坛搜集
主要除菌过滤器应该放在无菌区还是有菌区?
第七十五条 非最终灭菌产品的过滤除菌应当符合以下要 求: (一)可最终灭菌的产品不得以过滤除菌工艺替代最终灭 菌工艺。如果药品不能在其最终包装容器中灭菌,可用 0.22μm(更小或相同过滤效力)的除菌过滤器将药液 滤入预先灭菌的容器内。由于除菌过滤器不能将病毒或 支原体全部滤除,可采用热处理方法来弥补除菌过滤的 不足。
19:54
产品是否可以121℃湿热灭菌15分钟
否 是
产品是否可以湿热灭菌F0≥8 分钟,达到SAL≤10-6
否 是
灭菌工艺及设备验证

对灭菌过程中的温度、压力、时间等物理参数进行监 测和记录,确保符合工艺要求。
残留量检测
对灭菌后的产品进行残留物检测,确保无有毒有害物 质残留。
验证流程
实施验证
准备验证所需物品和资料
准备样品、试剂、仪器等,收集 相关工艺技术资料。
按照验证方案进行试验,记录各 项数据。
分析验证数据
对收集到的数据进行分析,评估 灭菌工艺的效果。
消毒验证方法
可以采用化学指示剂、生物指示剂等方法,对设备的消毒效果进行验证。同时,应关注消毒剂的选择和使用,避 免对设备造成腐蚀和损伤。
05
灭菌工艺与设备验证中 的问题与对策
问题一:灭菌不彻底
总结词
灭菌不彻底可能导致微生物残留,影响产品质量和安全性。
详细描述
灭菌不彻底的原因可能包括设备性能不佳、工艺参数设置不当、灭菌时间不足等 。为解决这一问题,需要定期对设备进行维护和校准,确保其性能稳定;同时, 优化工艺参数,如提高温度、延长灭菌时间等,以提高灭菌效果。
06
案例分析
案例一:某医院压力蒸汽灭菌设备的验证
01
02
03
验证目的
确保压力蒸汽灭菌设备性 能正常,能够达到预期的 灭菌效果。
验证方法
采用标准测试包,按照规 定程序进行灭菌,并对灭 菌后的物品进行生物指标 菌检测。
验证结果
经过多次验证,该压力蒸 汽灭菌设备性能稳定,灭 菌效果可靠,符合国家相 关标准和医院使用要求。
制定验证方案
明确验证目的、方法、范围和时 间安排等。
编写验证报告
根据验证结果编写报告,总结灭 菌工艺的有效性、可靠性和安全 性。
04
灭菌设备验证
设备性能验证
PDA TR26 除菌过滤验证(中文2008)

液体的除菌过滤PDA第26份技术报告(2008年修订本)制药科学与技术的PDA期刊增刊2008年第62卷,第S-5号1.0引言除菌过滤是从液体流中去除微生物*而对产品质量没有负面影响的过程。
(1-4)这份技术报告的目的是提供系统的方法,用于选择和验证液体除菌过滤应用的最适当过滤器。
PDA的第26份原始技术报告发表于1998年,标题为液体的除菌过滤,其中描述了一代制药科学家和工程师对除菌过滤的使用和验证。
由于过滤技术的加强以及制药行业近期产生了其他的法规要求,因而对原始报告进行了修改。
修订本涉及到了法规文件、标准以及科学出版物,其中包含更多的细节和支持性数据。
在20世纪60年代膜过滤器进入市场,当时认为0.45µm级别的膜为“除菌级”过滤器且成功应用于注射剂的除菌过滤。
使用serratia marcescens作为标准菌对这些过滤器进行确认,确认用于水质量测试的膜。
然而在1960年发布的论文中,美国FDA的Frances Bowman博士发现0.45µm的“除菌过滤的”培养基可受到一种生物的污染,在每平方厘米104-106以上的挑战水平下少量的这种生物可反复穿透0.45µm级别的膜。
(5)ASTM F838也由此产生,这是一种标准测试法,用于评估除菌级别的膜过滤器。
(6)在第6.4部分中对挑战生物进行了讨论。
1.1目的/ 适用范围工作组的主要目标是开发一份有关除菌过滤的科学的技术报告。
报告不会对区域的法规要求进行过多的描述,但是提供了最新的科学建议以供业内人士及制定除菌过滤政策的人员使用。
这份报告是一份指南性文件,其目的不是确立强制的除菌过滤标准。
报告中提出的概念与一些工艺有关,在这些工艺中除菌过滤器的性能是不可缺少的,而且这些概念不能通用于所有过滤工艺(例如,早期过滤或常规生物负载)。
这些概念包括但不限于细胞培养基、缓冲液、无菌工艺中的中间体暂存区、集中和最终无菌灌装。
除菌过滤工艺验证

除菌过滤工艺验证是一种用于确认过滤系统能够有效去除微生物的方法。
以下是一般的除菌过滤工艺验证的步骤:
设计验证方案:确定验证的目标、范围和计划。
考虑到产品的特性、要求和过滤系统的配置,制定验证方案。
准备验证样本:准备含有代表性微生物的验证样本。
可以使用经过标准化的微生物悬浮液或特定的微生物株系。
确保验证样本中微生物的浓度和存活率符合要求。
进行验证试验:将验证样本通过待验证的过滤系统进行过滤。
注意在试验过程中遵循严格的操作规程和规范,确保验证过程的准确性和可重复性。
收集样本和分析:收集通过过滤系统后的样本,并将样本送往实验室进行微生物分析。
通过分析验证样本中的微生物数量和存活率,评估过滤系统的除菌效果。
数据分析和结果评估:对验证试验的数据进行分析和解释,评估过滤系统的除菌效果。
与事先设定的标准或要求进行比较,确定是否符合要求。
编写验证报告:根据验证结果编写验证报告,详细记录验证方案、试验过程、数据分析和结论。
验证报告应清晰地说明过滤系统的除菌效果以及是否达到预期要求。
需要注意的是,除菌过滤工艺验证的具体方法和要求可能因不同的行业、产品和标准而有所不同。
在进行验证之前,应仔细研究适用的法规、行业指南和标准,并遵循相关的操作规程和规定。
此外,验证应由专业人员进行或在专业指导下进行,以确保验证过程的可靠性和有效性。
《化学药品注射剂灭菌无菌工艺研究及验证指导原则》

化学药品注射剂灭菌/无菌工艺研究及验证指导原则目录一、概述 (3)二、注射剂湿热灭菌工艺 (4)(一)湿热灭菌工艺的研究 (4)1.湿热灭菌工艺的确定依据 (4)2.微生物污染的监控 (7)(二)湿热灭菌工艺的验证 (9)1.物理确认 (9)2.生物学确认 (13)3.基于风险评估的验证方案设计 (16)三、注射剂无菌生产工艺 (16)(一)无菌生产工艺的研究 (16)1.除菌过滤工艺的研究 (16)2.无菌分装工艺的研究 (18)(二)无菌生产工艺的验证 (18)1.除菌过滤工艺验证 (19)2.无菌工艺模拟试验 (21)1/ 29四、附件 (24)五、参考文献 (27)2/ 291一、概述2无菌药品是指法定药品标准中列有无菌检查项目的制3剂和原料药,一般包括注射剂、无菌原料药及滴眼剂等。
4从严格意义上讲,无菌药品应不含任何活的微生物,但由5于目前检验手段的局限性,绝对无菌的概念不能适用于对6整批产品的无菌性评价,因此目前所使用的“无菌”概念,7是概率意义上的“无菌”。
特定批次药品的无菌特性只能通8过该批药品中活微生物存在的概率低至某个可接受的水平,即无菌保证水平(Sterility Assurance Level, SAL)来表征,910而这种概率意义上的无菌需通过合理设计和全面验证的灭11菌/除菌工艺过程、良好的无菌保证体系以及在生产过程中12执行严格的药品生产质量管理规范(GMP)予以保证。
13本指导原则主要参考国内外相关技术指导原则和标准14起草制订,重点对注射剂常用的灭菌/无菌工艺,即湿热灭15菌为主的终端灭菌工艺(terminal sterilizing process)和无16菌生产工艺(aseptic processing)的研究和验证进行阐述,17旨在促进现阶段化学药品注射剂的研究和评价工作的开展。
18本指导原则主要适用于无菌注射剂申请上市以及上市后变19更等注册申报过程中对灭菌/无菌工艺进行的研究和验证工作,相关仪器设备等的验证及常规再验证不包括在本指2021导原则的范围内。
2020版《化学药品注射剂灭菌无菌工艺研究及验证指导原则》
化学药品注射剂灭菌/无菌工艺研究及验证指导原则目录一、概述 (3)二、注射剂湿热灭菌工艺 (4)(一)湿热灭菌工艺的研究 (4)1.湿热灭菌工艺的确定依据 (4)2.微生物污染的监控 (7)(二)湿热灭菌工艺的验证 (9)1.物理确认 (9)2.生物学确认 (13)3.基于风险评估的验证方案设计 (16)三、注射剂无菌生产工艺 (16)(一)无菌生产工艺的研究 (16)1.除菌过滤工艺的研究 (16)2.无菌分装工艺的研究 (18)(二)无菌生产工艺的验证 (18)1.除菌过滤工艺验证 (19)2.无菌工艺模拟试验 (21)1/ 29四、附件 (24)五、参考文献 (27)2/ 291一、概述2无菌药品是指法定药品标准中列有无菌检查项目的制3剂和原料药,一般包括注射剂、无菌原料药及滴眼剂等。
4从严格意义上讲,无菌药品应不含任何活的微生物,但由5于目前检验手段的局限性,绝对无菌的概念不能适用于对6整批产品的无菌性评价,因此目前所使用的“无菌”概念,7是概率意义上的“无菌”。
特定批次药品的无菌特性只能通8过该批药品中活微生物存在的概率低至某个可接受的水平,即无菌保证水平(Sterility Assurance Level, SAL)来表征,910而这种概率意义上的无菌需通过合理设计和全面验证的灭11菌/除菌工艺过程、良好的无菌保证体系以及在生产过程中12执行严格的药品生产质量管理规范(GMP)予以保证。
13本指导原则主要参考国内外相关技术指导原则和标准14起草制订,重点对注射剂常用的灭菌/无菌工艺,即湿热灭15菌为主的终端灭菌工艺(terminal sterilizing process)和无16菌生产工艺(aseptic processing)的研究和验证进行阐述,17旨在促进现阶段化学药品注射剂的研究和评价工作的开展。
18本指导原则主要适用于无菌注射剂申请上市以及上市后变19更等注册申报过程中对灭菌/无菌工艺进行的研究和验证工作,相关仪器设备等的验证及常规再验证不包括在本指2021导原则的范围内。
除菌过滤工艺验证策略
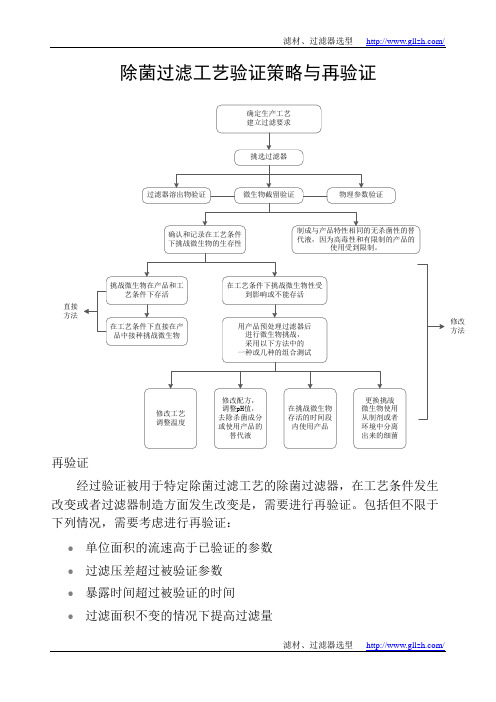
除菌过滤工艺验证策略与再验证再验证经过验证被用于特定除菌过滤工艺的除菌过滤器,在工艺条件发生改变或者过滤器制造方面发生改变是,需要进行再验证。
包括但不限于下列情况,需要考虑进行再验证:●单位面积的流速高于已验证的参数●过滤压差超过被验证参数●暴露时间超过被验证的时间●过滤面积不变的情况下提高过滤量●过滤温度改变●产品配方改变,包括浓度、pH或电导率●过滤器的灭菌程序改变●改变过滤器的制造商,或者过滤器的生产者改变了过滤膜的配方以及过滤器的其它结构性材料【实例分析】A. 修改的方法如前述,如果在工艺条件下挑战微生物不能存活或者生存力受到影响,在用产品预处理过滤器后,采用修改的方法进行测试。
下面给出一些方法修改的例子,实际应用时,可采用一种或几种进行组合测试。
应当认识到,如果有良好的科学解释,其它方式的方法修改也可能是适合的。
(1)减少暴露时间有些情况下,挑战微生物不一定在整个工艺时间下都能在产品中存活。
在挑战实验预处理阶段接近尾声的时候直接用足够浓度(见前文实施指导“D.培养基维护和挑战微生物制备”)的挑战微生物接种产品。
有一点是很重要的,将一个0.45微米阳性对照过滤器与测试过滤器平行运行,以确证微生物的尺寸和存活。
另一个办法是将挑战微生物在静态状态下置于挑战流体中。
将挑战微生物以工艺温度暴露在产品中,并在模拟工艺条件下用产品循环对过滤器进行预处理后,就可在最差条件(压差和流速)下挑战过滤器了。
挑战时间应被缩减,以确保在实际暴露、挑战和回收期间挑战微生物存活。
0.45微米阳性对照过滤器与测试过滤器平行运行,以确认微生物的存活。
(2)修改测试方法的参数在挑战有杀菌能力的产品时,测试方法参数修改是有用的,因为这种方法可以只改变一个工艺变量,例如温度。
使用这种方式以维持产品和细菌的相互反应。
这种方法不能解决所有可能的工艺/产品/挑战微生物的交互反应,但是可以实现标准挑战微生物的运用。
在实际工艺条件下使用产品对过滤器进行预处理后,工艺中的杀菌部分如温度被修改了,细菌挑战可以进行了。
《化学药品注射剂灭菌无菌工艺研究及验证指导原则》
化学药品注射剂灭菌/无菌工艺研究及验证指导原则目录一、概述 (3)二、注射剂湿热灭菌工艺 (4)(一)湿热灭菌工艺的研究 (4)1.湿热灭菌工艺的确定依据 (4)2.微生物污染的监控 (7)(二)湿热灭菌工艺的验证 (9)1.物理确认 (9)2.生物学确认 (13)3.基于风险评估的验证方案设计 (16)三、注射剂无菌生产工艺 (16)(一)无菌生产工艺的研究 (16)1.除菌过滤工艺的研究 (16)2.无菌分装工艺的研究 (18)(二)无菌生产工艺的验证 (18)1.除菌过滤工艺验证 (19)2.无菌工艺模拟试验 (21)1/ 29四、附件 (24)五、参考文献 (27)2/ 291一、概述2无菌药品是指法定药品标准中列有无菌检查项目的制3剂和原料药,一般包括注射剂、无菌原料药及滴眼剂等。
4从严格意义上讲,无菌药品应不含任何活的微生物,但由5于目前检验手段的局限性,绝对无菌的概念不能适用于对6整批产品的无菌性评价,因此目前所使用的“无菌”概念,7是概率意义上的“无菌”。
特定批次药品的无菌特性只能通8过该批药品中活微生物存在的概率低至某个可接受的水平,即无菌保证水平(Sterility Assurance Level, SAL)来表征,910而这种概率意义上的无菌需通过合理设计和全面验证的灭11菌/除菌工艺过程、良好的无菌保证体系以及在生产过程中12执行严格的药品生产质量管理规范(GMP)予以保证。
13本指导原则主要参考国内外相关技术指导原则和标准14起草制订,重点对注射剂常用的灭菌/无菌工艺,即湿热灭15菌为主的终端灭菌工艺(terminal sterilizing process)和无16菌生产工艺(aseptic processing)的研究和验证进行阐述,17旨在促进现阶段化学药品注射剂的研究和评价工作的开展。
18本指导原则主要适用于无菌注射剂申请上市以及上市后变19更等注册申报过程中对灭菌/无菌工艺进行的研究和验证工作,相关仪器设备等的验证及常规再验证不包括在本指2021导原则的范围内。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
3 Aseptic filtration Validation 除菌过滤验证
----生产管理者,质量管理者
下半部分:提高部分
4 Filtration process design 过滤系统的设计
---生产人员,生产管理者,工程人员
dun
认识我们使用的过滤器
亲水膜材质:
纤维素材料(NC) 尼龙(Nylon) 亲水聚偏二氟乙烯(PVDF) 聚谜砜(PES) 聚丙烯(PP)
疏水膜材质:
聚丙烯(PP) 疏水聚偏二氟乙烯(PVDF) 聚四氟乙烯(PTFE)
dun
第六步 制药工业中常用的三种滤器结构
dun
过滤器选择
1. 根据过滤对象 2. 根据亲水或疏水
3. 根据过滤阶段
Process step 生产步骤 Double bagging in class ISO 7 万级环境下双层包装(唯一编号) Packing including individual Quality Certificate (NC area) 每个独立包装中都有质量证书 Peel off label可剥离的标签
Filter integrity test operator certification 滤器操作者资格证书
Steritest school 无菌培训
灭菌过程设计,标准 Training 操作程序的培训, Education培训
surfaces
Extractables 析出物
Compatibility
12 10 8 6 4 2 0
0
Vmax
Traditional Flow Decay
10 20 30 40 50 60 time (min.)
dun
来自客户的问题
1 是不是选择 0.2um的过滤器就 能达到除菌的目的? 2 超滤可以达到除菌目的吗? 3 为什么不同步骤选择不同类型 的过滤器?不同操作压力? 4 不同材质对过滤效果为什么有 影响?
Objective目的 Deliver the highest quality standard product 提供符合最高质量标准的产品
dun
dunSection 2: Introduction to Filtration
第三步:根据过滤阶段 —除菌过滤膜
坚强,硬不易碎,更薄 曲折的通道 65%-75%开孔率 大小排除-颗粒截流与流速和压力无关 无菌过滤器必须具有大于99.99999%截流
dun
5
Agenda 目录
1 Filter selection 过滤器的应用与选择
----采购人员,生产管理者,工程人员
上半部分:基础部分
2 Integrity testing 完整性测试
----生产人员,生产管理者,质量管理者,工程人员
3 Aseptic filtration Validation 除菌过滤验证
dun
dunSection 2: Introduction to Filtration
3
第三步:根据过滤阶段 ----预过滤
可以给出公称空径,均匀 较薄(小于1毫米),较小吸附 给出颗粒减少的比例(95-99.9%) 例如-纤维素酯涂纤维或聚酯片 多空聚合物铸造而成,可以控制孔 径,均匀的多,可以预测截留情况
dun
4
第四步:选择孔径
澄清过滤—深层过滤膜
0.1um,0.3um,0.5um,1.0um,3.0um,10um,25um,50um,1 00um,
预过滤—表面过滤膜
0.2um,0.2+0.5um, 0.5+1.2um,
除菌过滤—绝对过滤膜
0.1um,0.22um,0.45um,
dun
第五步:选择过滤膜材质
除菌级过滤器 (47 mm Durapore
0.22 µm disc)
Waste
Incubate 7 days, Inspect for Growth 培养7天,观察有无菌生长
dunSection 3: Integrity Testing Theory
1
非破坏性完整性测试?
大于0.22um的缺陷孔 微孔滤膜
integrity
Test过滤器的完
整性检测
Product
specific
产品完整性测
试,标准及 方
Tester
法
IQ OQ检测仪的安装
运行确认
2完整性测试
Agenda 目录
1 Filter selection 过滤器的应用与选择
----采购人员,生产管理者,工程人员
上半部分:基础部分
2 Integrity testing 完整性测试
1
5
0.22 µm 4
4. 选择孔径
5. 选择材质
23
6. 选择过滤器形式
7. 选择过滤面积
dun
dunSection 2: Introduction to Filtration
t/V (min/L)
第七步:过滤面积确定方法加速衰减法(Vmax)
收集10分钟内,特定压力下过滤数据 Cumulative volume (V) 累积体积 Time (t) 时间
dun
Millipore 不对称聚醚砜过滤器
新设计的聚醚砜过滤器 化学兼容性 1<pH<14
所用膜是复合型的,非对称结构的 流速快(过滤缓冲液,消毒剂和腐蚀性液体)
是满足ASTM 838-83标准的除菌级过滤器(107 B.diminuta/cm²) 具有验证的性能
依据PDA标准执行验证
dun
最终包装
大容量注射剂
预过滤
预过滤 : Milligard (CWSS)
除菌过滤
最终过滤 : 最终过滤 Durapore HL (0.45 µm) 或SHF(0.22 µm)
API原料药
N F气F - S体te除rili菌zation
N澄FF清- B过ul滤k C la rific a tio n
N C
Fl预aFr -i过f Sicoa滤ltvioenn
dun
dunSection 2: Introduction to Filtration
除菌过滤 在无菌药品生产中的应用
灭菌设备
清洗 设备
干热 灭菌
蒸汽 灭菌
消毒剂
高风险操作区域 混合 灌装 其他
VHP,EO 发生器
冻干机
最终 灭菌
水预 处理
纯蒸汽 发生器
压缩空气
无菌检测
8
第一步:选择过滤对象
反渗透
超滤
t
种子SFee培rem养d entor
F发erm e酵ntor
N预FF过- 滤 C lar ific a t io n
NC预FlaFr过-ific滤a tion
SE有ox机tlvrae溶cnt剂tio萃n 取
层 析/活性碳/溶剂沉淀
TFFCo切nc向en流tr浓a ti缩on
除NS tF菌eFr -il过iz a滤ti o n
应该在什么时候做完整性测试?
PDA技术报告26号 一般认为过滤前后的常规性完整性检测是现行GMP的要求. EU GMP 对于已灭菌的滤器的完整性检测应该在使用前和使用后立即执行,例如泡点,
扩散流及压力保持试验. 中国GMP 滤器在使用后以及在使用前,应有书面的完整性试验纪录。这需要使用一些
0.22um的意义
制造工艺
生产环境 (Class 100.000十万级环境) (Class 10.000) 最终装配,检测及包装
完整性检测 100%完整性检测
生产技术与控制 全自动 无缝结合 温差实验及细菌挑战
dun
Durapore Manufacturing Quality Control Release Tests
起泡点测试,扩散测试
制造商及使用者ຫໍສະໝຸດ dun除菌过滤器的定义是什么?
Defined by ASTM F838-83 (1993) 给出定义 A filter which, when challenged with the bacterium Brevundimonas diminuta, at a minimum concentration of 107 cfu per cm2 of filter surface area, will produce a sterile effluent. 除菌级过滤器是指符合以下标准的过滤器:用每平方 厘米107CFu的缺陷性假单胞菌进行挑战这一滤器, 下游液体无菌。
起泡点测试原理
压缩空气
σ
P2
d
θ Water
压缩空气
dun
dun
手工测试起泡点的方法
冲洗
加压
增加压力直到连续起泡在下游出现
dun
扩散流检测原理
P1 Air
σ
P2
d
θ Water
施加压力
L L
P
A
P0
dun
手工测试扩散流方法
压缩空气
上游压力
起泡点、扩散流的合格标准是否具有意义?
破坏性和非破坏性测试 两者必须建立关联
dun
为什么对除菌过滤器要作完整性测试?
无菌工艺及验证需要:
确认正确的过滤孔径 检测O形环,垫圈,密封垫的泄漏 确认蒸汽和消毒锅灭菌后完整性
法规要求及贸易需要
出口认证(FDA,COS, EDMF) SDA检查
dun
完整性测试的方法有哪些?
分类 破坏性
测试名称 金标准 测试实施者
细菌挑战测试
制造商
非破坏性
下游监测气流速度 dun
dunSection 3: Integrity Testing Theory