Organic Syntheses, Coll. Vol. 5, p.277 (1973); Vol. 48, p.56 (1968).
靛红的合成
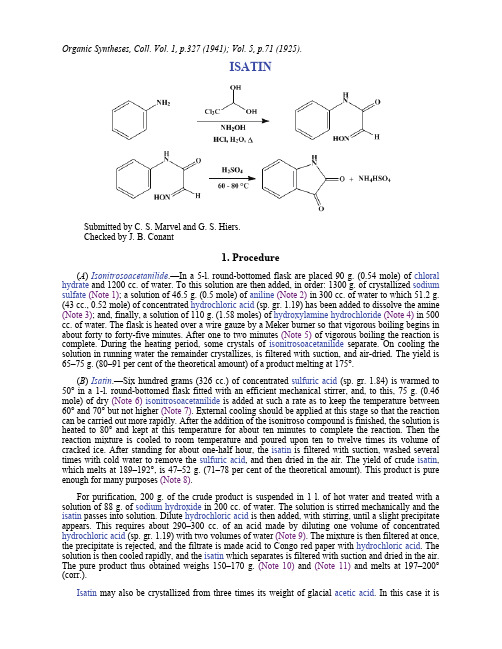
Organic Syntheses, Coll. Vol. 1, p.327 (1941); Vol. 5, p.71 (1925).ISATINSubmitted by C. S. Marvel and G. S. Hiers.Checked by J. B. Conant1. Procedure(A) Isonitrosoacetanilide.—In a 5-l. round-bottomed flask are placed 90 g. (0.54 mole) of chloral hydrate and 1200 cc. of water. To this solution are then added, in order: 1300 g. of crystallized sodium sulfate(Note 1); a solution of 46.5 g. (0.5 mole) of aniline(Note 2) in 300 cc. of water to which 51.2 g.(43 cc., 0.52 mole) of concentrated hydrochloric acid (sp. gr. 1.19) has been added to dissolve the amine (Note 3); and, finally, a solution of 110 g. (1.58 moles) of hydroxylamine hydrochloride(Note 4) in 500 cc. of water. The flask is heated over a wire gauze by a Meker burner so that vigorous boiling begins in about forty to forty-five minutes. After one to two minutes (Note 5) of vigorous boiling the reaction is complete. During the heating period, some crystals of isonitrosoacetanilide separate. On cooling the solution in running water the remainder crystallizes, is filtered with suction, and air-dried. The yield is 65–75 g. (80–91 per cent of the theoretical amount) of a product melting at 175°.(B) Isatin.—Six hundred grams (326 cc.) of concentrated sulfuric acid (sp. gr. 1.84) is warmed to 50° in a 1-l. round-bottomed flask fitted with an efficient mechanical stirrer, and, to this, 75 g. (0.46 mole) of dry (Note 6)isonitrosoacetanilide is added at such a rate as to keep the temperature between 60° and 70° but not higher (Note 7). External cooling should be applied at this stage so that the reaction can be carried out more rapidly. After the addition of the isonitroso compound is finished, the solution is heated to 80° and kept at this temperature for about ten minutes to complete the reaction. Then the reaction mixture is cooled to room temperature and poured upon ten to twelve times its volume of cracked ice. After standing for about one-half hour, the isatin is filtered with suction, washed several times with cold water to remove the sulfuric acid, and then dried in the air. The yield of crude isatin, which melts at 189–192°, is 47–52 g. (71–78 per cent of the theoretical amount). This product is pure enough for many purposes (Note 8).For purification, 200 g. of the crude product is suspended in 1 l. of hot water and treated with a solution of 88 g. of sodium hydroxide in 200 cc. of water. The solution is stirred mechanically and the isatin passes into solution. Dilute hydrochloric acid is then added, with stirring, until a slight precipitate appears. This requires about 290–300 cc. of an acid made by diluting one volume of concentrated hydrochloric acid (sp. gr. 1.19) with two volumes of water (Note 9). The mixture is then filtered at once, the precipitate is rejected, and the filtrate is made acid to Congo red paper with hydrochloric acid. The solution is then cooled rapidly, and the isatin which separates is filtered with suction and dried in the air. The pure product thus obtained weighs 150–170 g. (Note 10) and (Note 11) and melts at 197–200° (corr.).Isatin may also be crystallized from three times its weight of glacial acetic acid. In this case it isobtained in large brown-red crystals which melt at 196–197°.2. Notes1. Several runs were made in which the amounts of water and sodium sulfate were varied over a considerable range, and this concentration was found to give the best yield of product of good quality. The sodium sulfate seems to have more than a salting-out effect. If a saturated solution of sodium chloride is used no product is obtained.2. Redistilled aniline boiling over a 2° range was used in these experiments. The ordinary "pure" grade gives slightly lower yields.3. If the aniline is not in solution, a considerable quantity of tarry material is formed during the heating period. No tar is formed when the method described is used.4. The hydroxylamine hydrochloride used was the crude material prepared as described on p. 318. Preliminary experiments showed that this reagent must be present in considerable excess. Equally good results were obtained by using a solution of crude hydroxylamine sulfate which also contained sodium sulfate and ammonium sulfate with a little excess sulfuric acid. The hydroxylamine content was determined in this solution by titration with potassium permanganate solution. When this crude solution is used, the addition of sodium sulfate is not always necessary.5. Longer heating of the reaction mixture gives a lower yield of dark-colored product.6. If too much moisture is left in the isonitrosoacetanilide it is not easy to control the reaction with sulfuric acid.7. The reaction does not start below 45–50° but becomes too violent above 75–80°. If the temperature becomes too high, the entire run is lost by charring. Stirring is needed to prevent local overheating.8. In some smaller preparations when the sulfuric acid solution was poured on ice a yellow compound precipitated, which was shown to be the oxime of isatin. It has also been isolated from the acid mother liquors from which the isatin has separated. The oxime probably owes its formation to the hydrolysis of some unaltered isonitrosoacetanilide (J. P. Wibaut,1 private communication).9. The correct amount of acid that must be added to precipitate the impurities but not the isatin will vary with different samples of crude isatin. If too much acid is added, some isatin comes down with the impurities. This may be saved and added to a subsequent run.10. The yield of isatin is lower than for some of its derivatives. The explanation given in the literature is that some sulfonation occurs during the treatment with sulfuric acid, with corresponding loss of product.11. This method can be applied successfully to other isatin derivatives. Thus, under the same conditions, 54 g. of p-toluidine gives 75–77 g. (83–86 per cent of the theoretical amount) of isonitrosoaceto-p-toluidine melting at 162°. Eighty grams of this isonitroso compound treated as described under isonitrosoacetanilide gives 65–68 g. (90–94 per cent of the theoretical amount) of crude 5-methyl isatin melting at 179–183°. This is purified as described under isatin by solution in sodium hydroxide and partial neutralization to throw out the impurities or by recrystallization from three parts of glacial acetic acid. The purified 5-methyl isatin melts at 187°.3. DiscussionIsatin can be prepared by the oxidation of indigo;2 and the condensation of aniline, chloral hydrate, and hydroxylamine salts, followed by the action of sulfuric acid.3 The latter method by Sandmeyer3 seemed most promising and has been studied in detail. The procedure described differs from it in the use of hydroxylamine hydrochloride itself instead of a crude solution of hydroxylamine sulfate, and in the use of sodium sulfate to salt out the isonitroso compound.This preparation is referenced from:z Org. Syn. Coll. Vol. 5, 635References and Notes1.Wibaut and Geerling, Rec. trav. chim. 50, 41 (1931).2.Erdmann, J. prakt. Chem. 24, 11 (1841); Laurent, ibid. 25, 434 (1842); Gericke, ibid. 95, 177(1865); Knop, ibid. 97, 86 (1866); Knape, ibid. (2) 43, 211 (1891); Hofmann, Ann. 53, 10 (1845); Sommaruga, Ann. 190, 369 (1878); Gericke, Jahresber. 580 (1865); Forrer, Ber. 17, 976 (1884); Diez and Co., Ger. pat. 229,815 [Frdl. 10, 353 (1910–12)]; Rabinovich and Dzirkal, Khim. Farm. Prom. 1933, 190 [C. A. 28, 475 (1934)]; Henesy, J. Soc. Dyers Colourists, 53, 345, 347 (1937).3.Sandmeyer, Helvetica Chim. Acta 2, 237, 239 (1919); Geigy, Brit. pat. 128,122 [C. A. 13, 2375(1919)].AppendixChemical Abstracts Nomenclature (Collective Index Number);(Registry Number)isonitroso compoundindigohydroxylamine saltssulfuric acid (7664-93-9)hydrochloric acid (7647-01-0)acetic acid (64-19-7)aniline (62-53-3)sodium hydroxide (1310-73-2)potassium permanganate (7722-64-7)sodium chloride (7647-14-5)sodium sulfate (7757-82-6)Hydroxylamine hydrochloride (5470-11-1)ammonium sulfate (7783-20-2)hydroxylamine (7803-49-8)Isatin (91-56-5)chloral hydrate (302-17-0)isonitrosoacetanilide (1769-41-1)hydroxylamine sulfate (10046-00-1)5-methyl isatin(608-05-9)p-toluidine (106-49-0)isonitrosoaceto-p-toluidine Copyright © 1921-2005, Organic Syntheses, Inc. All Rights Reserved。
Organic Syntheses, Coll. Vol. 5, p.157 (1973); Vol. 44, p.15 (1964).

Organic Syntheses, Coll. Vol. 5, p.157 (1973); Vol. 44, p.15 (1964).t-BUTYL AZIDOFORMATE[Formic acid, azido-, tert-butyl ester]Submitted by Louis A. Carpino, Barbara A. Carpino, Paul J. Crowley, Chester A. Giza, and PaulH. Terry1.Checked by Virgil Boekelheide and S. J. Cross.1. ProcedureIn a 1-l. round-bottomed flask fitted with a mechanical stirrer are placed 82 g. (0.62 mole) of t-butyl carbazate,2 72 g. of glacial acetic acid, and 100 ml. of water. The solution is cooled in an ice bath, and 47.0 g. (0.68 mole) of solid sodium nitrite is added over a period of 40–50 minutes, the temperature being kept at 10–15° (Note 1). The mixture is allowed to stand in the ice bath for 30 minutes, 100 ml. of water is added, and the floating oil is extracted into four 40-ml. portions of ether. The combined ether extracts are washed twice with 50-ml. portions of water and with 40-ml. portions of 1M sodium bicarbonate solution until no longer acidic (about three washings are required). The solution is dried over magnesium sulfate, and the ether is removed by distillation from a water bath maintained at 40–45°; water aspirator pressure of 140–150 mm. is used. The pressure is then lowered to 70 mm., and the water bath temperature is raised to 90–95°. The azide is distilled (Caution! (Note 2)) using a Claisen flask and is collected at 73–74° (70 mm.), n24D 1.4227, after a few drops of fore-run. The yield is 57–72.8 g. (64–82%) (Note 3) and (Note 4).2. Notes1. The sodium nitrite may be added as a concentrated aqueous solution.2. It is recommended that the distillation be carried out behind a safety shield. The submitters have distilled this compound several hundred times without incident under the conditions given on a scale up to 300–400 g. per run. On the other hand, Prof. P. G. Katsoyannis (University of Pittsburgh School of Medicine, Pittsburgh, Pennsylvania) has reported that an explosion took place in the receiving flask while the compound was being distilled under conditions previously used without incident. The reason for the explosion could not be traced. According to Prof. R. Schwyzer (Ciba, Ltd., Basel, Switzerland) tests at a Swiss Federal Institute showed that the compound could not be exploded by mere heating: it simply decomposes. For explosion, one must apply a primary explosive such as lead azide or silver azide. An attempt by the submitters to distil the azide at atmospheric pressure resulted in vigorous carbonization, but no explosion occurred. In view of the potential hazard some investigators prefer not to distil the azide; they use the crude material after removal of solvent. High yields of carbo-t-butoxy derivatives may be obtained in this way.3. When freshly distilled, the azide is water-white. When the azide is allowed to stand for several weeks, it slowly develops a light yellow color; however, this does not appear to affect its reactivity as an acylating agent.34. The azide should be handled with adequate ventilation. Careless inhalation of the substance was accompanied by development of a painful throbbing headache or a sensation of giddiness or both. These effects disappeared within several hours upon exposure to fresh air.3. Discussiont-Butyl azidoformate has been prepared by a variety of procedures,3,4,5,6,7,8,9,10,11,12 of which the present procedure and that described elsewhere in this series12appear most satisfactory.Because of the instability of t-butyl chloroformate a number of carbonic acid derivatives have been prepared and studied as reagents for the introduction of the carbo-t-butoxy group. A listing of these reagents and references to their preparation may be found in reference 13. In spite of some disadvantages the most widely used reagent is still t-butyl azidoformate, although t-butyl 2,4,5-trichlorophenyl-carbonate appears to be another potentially useful reagent. t-Butyl azidoformate is a convenient reagent for the acylation of amines, hydrazines, and similar compounds.3The acylation product of hydroxylamine, t-butyl N-hydroxycarbamate,5 is a valuable intermediate in the synthesis of O-substituted hydroxylamines such as O-acyl- and O-sulfonylhydroxylamines, many of which are valuable aminating agents and have not be obtained in any other way.14,15 This preparation is referenced from:z Org. Syn. Coll. Vol. 5, 160z Org. Syn. Coll. Vol. 6, 199z Org. Syn. Coll. Vol. 6, 203z Org. Syn. Coll. Vol. 6, 207z Org. Syn. Coll. Vol. 6, 418z Org. Syn. Coll. Vol. 7, 70References and Notes1.Department of Chemistry, University of Massachusetts, Amherst, Massachusetts.2.L. A. Carpino, D. Collins, and S. Göwecke, this volume, p. 166.3.L. A. Carpino, J. Am. Chem. Soc., 79, 4427 (1957).4.L. A. Carpino, J. Am. Chem. Soc., 79, 98 (1957).5.L. A. Carpino, C. A. Giza, and B. A. Carpino, J. Am. Chem. Soc., 81, 955 (1959).6.K. P. Polzhofer, Chimia (Aarau), 23, 298 (1969).7.H. Yajima and H. Kawatani, Chem. Pharm. Bull. (Tokyo), 16, 182 (1968).8.M. Itoh and D. Morino, Experientia, 24, 101 (1968).9.Y. A. Kiryushkin and A. I. Miroshnikov, Experientia, 21, 418 (1965).10.K. Inouye, M. Kanayama, and H. Otsuka, Nippon Kagaku Zasshi, 85, 599 (1964).11. D. S. Tarbell, Accounts Chem. Res., 2, 296 (1969).12.M. A. Insalaco and D. S. Tarbell, Org. Syntheses, 50, 9 (1970).13.L. A. Carpino, K. N. Parameswaran, R. K. Kirkley, J. W. Spiewak, and E. Schmitz, J. Org.Chem., 35, 3291 (1970).14.L. A. Carpino, J. Am. Chem. Soc., 82, 3133 (1960).15.L. A. Carpino, J. Am. Chem. Soc., 85, 2144 (1963).AppendixChemical Abstracts Nomenclature (Collective Index Number);(Registry Number)acetic acid (64-19-7)ether (60-29-7)sodium bicarbonate (144-55-8)sodium nitrite(7632-00-0)hydroxylamine (7803-49-8)magnesium sulfate (7487-88-9)silver azidet-BUTYL AZIDOFORMATE,Formic acid, azido-, tert-butyl ester (1070-19-5)t-butyl carbazate (870-46-2)t-butyl chloroformatet-butyl 2,4,5-trichlorophenyl-carbonate (16965-08-5)t-butyl N-hydroxycarbamate (36016-38-3)lead azideCopyright © 1921-2005, Organic Syntheses, Inc. All Rights Reserved。
钯碳 水合肼 还原 硝基

Organic Syntheses, Coll. Vol. 5, p.30 (1973); Vol. 40, p.5 (1960).2-AMINOFLUORENE[2-Flurenylamine]Submitted by P. M. G. Bavin1Checked by John C. Sheehan and Roger E. Chandler..1. ProcedureIn a 2-l. three-necked round-bottomed flask, equipped with a mechanical stirrer (Note 1), reflux condenser, and dropping funnel, are placed 30 g. of pure 2-nitrofluorene, m.p. 157° [Org. Syntheses, Coll. Vol. 2, 447 (1943)], and 250 ml. of 95% ethanol. After warming to 50° on a steam bath, 0.1 g. of palladized charcoal catalyst (previously moistened with alcohol) is added (Note 2) and the stirrer is started. About 15 ml. of hydrazine hydrate is added from the dropping funnel during 30 minutes (Note 3). At this point an additional 0.1 g. of catalyst (previously moistened with alcohol) is added and the mixture is heated until the alcohol refluxes gently. After 1 hour the nitrofluorene has dissolved completely and the supernatant liquor is almost colorless.The catalyst is removed by filtration with gentle suction through a thin layer of Celite (Note 4). The flask is rinsed with 30 ml. of hot alcohol which is then used to wash the catalyst and Celite. The combined filtrates are concentrated under reduced pressure to about 50 ml. (Note 5) and then heated to boiling at atmospheric pressure. When 250 ml. of hot water is added slowly, 2-aminofluorene is precipitated as a colorless, crystalline powder. After cooling in an ice bath, the 2-aminofluorene is collected, washed with water, and dried in the dark in a vacuum desiccator. The product melts at 127.8–128.8° (Note 6) and amounts to 24–25 g. (93–96%).2. Notes1. If the stirring is omitted, the nitrofluorene takes longer to dissolve.2. A suitable catalyst is 10% palladium-on-charcoal, such as is supplied by Baker and Company, Inc., 113 Astor Street, Newark 5, New Jersey.3. The reaction is exothermic, and too rapid addition of the hydrazine may cause the mixture to foam out of the condenser.4. Caution! The catalyst is often pyrophoric and should be kept moistened with alcohol. Celite is a diatomaceous earth filter aid.5. A rotary evaporator is very convenient for the concentration since some of the amine invariably crystallizes toward the end.6. The melting point is that reported in Organic Syntheses, Coll. Vol. 2, 448 (1943), for a recrystallized sample.3. DiscussionThe preparation of 2-aminofluorene reported previously in Organic Syntheses[Coll. Vol. 2, 448 (1943)] we based on the method of Diels.2The present procedure illustrates a general method for the reduction of aromatic nitro compounds to aromatic amines using hydrazine and a hydrogenation catalyst such as palladium, platinum, nickel, iron, or rethenium. The literature on this procedure up to 1963 has been reviewed.3 In many instances the catalytic hydrazine reductions give yields of amine equal to or better than those obtained by directcatalytic hydrogenation or other reduction methods. Both the apparatus and the procedure are simple. Under appropriate conditions the method may be used for the dehalogenation of aliphatic and aromatic halides,3 a reaction for which palladium appears to be a specific catalyst. The method has also been used for the reduction of azobenzene and azoxybenzene to hydrazobenzene (80–90%),4 as well as for the synthesis of steroid aziridines by reduction of mesylate esters by vicinal azido alcohols (using Raney nickel).5References and Notes1.National Research Council of Canada Post-doctorate Fellow, 1954-56, at the University ofOttawa, Ottawa, Ontario.2.O. Diels, Ber., 34, 1758 (1901).3. A. Furst, R. C. Berlo, and S. Hooton, Chem. Rev., 65, 51 (1965).4.P. M. G. Bavin, Can. J. Chem., 36, 238 (1958).5.K. Ponsold, Ber., 97, 3524 (1964).AppendixChemical Abstracts Nomenclature (Collective Index Number);(Registry Number)2-Flurenylaminepalladized charcoal catalystpalladium-on-charcoalethanol (64-17-5)iron (7439-89-6)platinum (7440-06-4)nickel,Raney nickel (7440-02-0)palladium (7440-05-3)hydrazine hydrate (7803-57-8)hydrazine (302-01-2)Azoxybenzene (495-48-7)Azobenzene (103-33-3)2-Nitrofluorene (607-57-8)2-Aminofluorene(153-78-6)Nitrofluoreneretheniumhydrazobenzene (122-66-7) Copyright © 1921-2005, Organic Syntheses, Inc. All Rights Reserved。
重氮化反应 氨基变卤素

Organic Syntheses, Coll. Vol. 2, p.351 (1943); Vol. 19, p.55 (1939).IODOBENZENE[Benzene, iodo-]Submitted by H. J. Lucas and E. R. Kennedy.Checked by John R. Johnson and P. L. Barrick.1. ProcedureIn a 3- or 5-gallon stoneware crock are placed 950 cc. (1130 g., 11.7 moles) of concentrated hydrochloric acid (sp. gr. 1.19), 950 cc. of water, 200 g. (196 cc., 2.15 moles) of aniline, and 2 kg. of ice (Note 1). The mixture is agitated by a mechanical stirrer, and, as soon as the temperature drops below 5°, a chilled solution of 156 g. (2.26 moles) of sodium nitrite in a measured volume (700–1000 cc.) of water is introduced fairly rapidly from a separatory funnel, the stem of which projects below the surface of the reaction mixture. The addition should not be fast enough to cause the temperature to rise to 10° or to cause evolution of oxides of nitrogen. The last 5 per cent of the nitrite solution is added more slowly, and the reaction mixture is tested with starch-iodide paper at intervals until an excess of nitrous acid is indicated.Stirring is continued for ten minutes, and if necessary the solution is filtered rapidly through a loose cotton plug in a large funnel. An aqueous solution of 358 g. (2.16 moles) of potassium iodide is added and the reaction mixture allowed to stand overnight. The mixture is transferred to a large flask (or two smaller flasks) and heated on a steam bath, using an air-cooled reflux condenser, until no more gas is evolved, then allowed to cool and stand undisturbed until the heavy organic layer has settled thoroughly.A large part of the upper aqueous layer is siphoned off, and discarded (Note 2). The residual aqueous and organic layers are made alkaline by the cautious addition of strong sodium hydroxide solution (100–125 g. of solid technical sodium hydroxide is usually required) and steam-distilled at once. The last one-third of the steam distillate is collected separately and combined with the aqueous layer separated from the earlier portions of the distillate. This mixture is acidified with 5–10 cc. of concentrated sulfuric acid and steam-distilled again. The iodobenzene from this operation is combined with the main portion and dried with 10–15 g. of calcium chloride(Note 3) and (Note 4). Distillation under reduced pressure gives 327–335 g. (74–76 per cent of the theoretical amount) of iodobenzene, b.p. 77–78°/20 mm. or 63–64°/8 mm. (Note 5).2. Notes1. If more ice is used a portion remains unmelted after the diazotization is completed.2. If a good separation has been made not more than 1–2 g. of iodobenzene is lost with the upper layer.3. An appreciable amount of iodobenzene is retained by the solid calcium chloride. By treating the spent drying agent with water 8–12 g. of iodobenzene can be recovered.4. The crude iodobenzene weighs 350–355 g. (80 per cent of the theoretical amount) and is pure enough for many purposes without redistillation.5. If the distillation is carried too far, the distillate will be colored.3. DiscussionThe preparation of iodobenzene by iodination of benzene, with iodine and nitric acid, and a survey of preparative methods have been given in an earlier volume.1 The present procedure, based upon the method of Gattermann,2 gives a purer product.This preparation is referenced from:z Org. Syn. Coll. Vol. 5, 660z Org. Syn. Coll. Vol. 5, 665References and Notes. Syn. Coll. Vol. I, 1941, 323.2.Gattermann-Wieland, "Laboratory Methods of Organic Chemistry," p. 283. Translated from thetwenty-fourth German edition by W. McCartney, The Macmillan Company, New York, 1937.AppendixChemical Abstracts Nomenclature (Collective Index Number);(Registry Number)oxides of nitrogencalcium chloride (10043-52-4)sulfuric acid (7664-93-9)hydrochloric acid (7647-01-0)Benzene (71-43-2)aniline (62-53-3)sodium hydroxide (1310-73-2)nitric acid (7697-37-2)potassium iodide (7681-11-0)sodium nitrite (7632-00-0)nitrous acid (7782-77-6)iodine (7553-56-2)Iodobenzene,Benzene, iodo-(591-50-4)Copyright © 1921-2005, Organic Syntheses, Inc. All Rights Reserved。
Organic Syntheses有机合成

Organic Syntheses(有机合成手册), John Wiley & Sons (免费)/Named Organic Reactions Collection from the University ofOxford (有机合成中的命名反应库) (免费)http://www.chem.ox.ac. ... rganicReac...有机化学资源导航Organic Chemistry Resources Worldwide/有机合成文献综述数据库Synthesis Reviews (免费)/srev/srev.htmCAMEO (预测有机化学反应产物的软件)http://zarbi.chem.yale ... o/index.shtmlCarbohydrate Letters (免费,摘要)/Carbohydrate_Letters/Carbohydrate Research (免费,摘要)/locate/carresCurrent Organic Chemistry (免费,摘要)/coc/index.htmlElectronic Encyclopedia of Reagents for Organic Synthesis (有机合成试剂百科全书e-EROS)/eros/European Journal of Organic Chemistry (免费,摘要)http://www.interscienc ... es/1434-193X/Methods in Organic Synthesis (MOS,有机合成方法)/is/database/mosabou.htmOrganic Letters (免费,目录)/jo ... f7/index.htmlOrganometallics (免费,目录)/jo ... d7/index.htmlRussian Journal of Bioorganic Chemistry (Bioorganicheskaya Khimiya) (免费,摘要) http://www.wkap.nl/journalhome.htm/1068-1620Russian Journal of Organic Chemistry (Zhurnal Organicheskoi Khimii) (免费,摘要) http://www.maik.rssi.ru/journals/orgchem.htmScience of Synthesis: Houben-Weyl Methods of Molecular Transformation/Solid-Phase Synthesis database (固相有机合成)/chem_db/sps.htmlSynthetic Communications (免费,摘要)/ ... productid/SCCSyntheticPages (合成化学数据库) (免费)/The Complex Carbohydrate Research Center (复杂碳水化合物研究中心)/合成材料老化与应用(免费,目录)http://hccllhyyy.perio ... /default.html金属卡宾络合物催化的烯烃复分解反应(免费) ... 02/2.6%20.htm上海化学试剂研究所/英国化学数据服务中心CDS (Chemical Database Service)/cds/cds.html英国皇家化学会碳水化合物研究组织(Carbohydrate Group of the Royal Society of Chemistry)/lap/rsccom/dab/perk002.htm有机反应催化学会(ORCS, Organic Reaction Catalysis Society)/有机合成练习(免费)/中国科学院成都有机化学研究所:催化与环境工程研究发展中心/MainIndex.htm金属有机及元素有机化学:CASREACT - Chemical Reactions Database(CAS的化学反应数据库)/CASFILES/casreact.html日本丰桥大学Jinno实验室的研究数据库(液相色谱、多环芳烃/药物/杀虫剂的紫外谱、物性) (免费)http://chrom.tutms.tut ... H/research...A New Framework for Porous Chemistry (金属有机骨架) (免费) ... 83432324.htmlActa Crystallographica Section B (免费,摘要)http://journals.iucr.o ... homepage.htmlActa Crystallographica Section E (免费,摘要)http://journals.iucr.o ... homepage.htmlBibliographic Notebooks for Organometallic Chemistryhttp://www.ensc-lille. ... bco/bnoc.htmlBiological Trace Element Research (生物痕量元素研究杂志) (免费,摘要)http://www.humanapress ... =0163-4984...Journal of Organometallic Chemistry (免费,摘要)/locate/jnlabr/jomOrganic Letters (免费,目录)/jo ... f7/index.htmlOrganometallics (免费,目录)/jo ... d7/index.htmlSyntheticPages (合成化学数据库) (免费)/金属卡宾络合物催化的烯烃复分解反应(免费) ... 02/2.6%20.htm金属有机参考读物:The Organometallic HyperTextBook by Rob Toreki/organomet/index.html金属有机化学国家重点实验室,中国科学院上海有机所/元素有机化学国家重点实验室(南开大学)/在线网络课程:有机金属反应和均相催化机理(Dermot O'Hare 主讲)http://www.chem.ox.ac. ... ot/organomet/药物化学:Fisher Scientific/PubMed: MEDLINE和PREMEDLINE (免费)/PubMed/生物医药:BioMedNet: The World Wide Club for the Biological and Medical Community /AIDSDRUGS (艾滋病药物) (免费) ... aidsinfs.htmlautodock (分子对接软件) (免费) ... doc/autodock/DIRLINE (卫生与生物医药信息源库) (免费)/HISTLINE (医药史库) (免费)/TOXNET (化合物毒性相关数据库系列) (免费)/日本药典,第14版(免费)http://jpdb.nihs.go.jp/jp14e/index.html小分子生物活性数据库ChemBank (免费)/Ashley Abstracts Database (药物研发、市场文献摘要) (免费) ... ey/search.aspBIOSIS/BIOSIS/ONLINE/DBSS/biosisss.html从检索药物交易信息库PharmaDeals (部分免费)/从ChemWeb检索有机药物用途及别名库Negwer: organic-chemical drugs and their synonyms (部分免费)http://cwgen.chemweb.c ... ersearch.html美国常用药品索引库RxList (免费)/美国国家医学图书馆NLM的免费在线数据库(免费)/ho ... internet.html制药公司目录(Pharmaceutical Companies on Virtual Library: Pharmacy Page)/company.html37℃医学网/AAPS PharmSci (免费,全文)/Abcam Ltd.有关抗体、试剂的销售,抗体的搜索)/Acta Pharmaceutica (免费,摘要)http://public.srce.hr/acphee/Advanced Drug Delivery Reviews (免费,摘要)http://www.elsevier.nl/locate/drugdelivAmerican Journal of Drug and Alcohol Abuse (免费,摘要)/ ... productid/ADAAmerican Journal of Pharmaceutical Education (AJPE) (免费,全文)/Amgen Inc. (医药)/Anita's web picks (药学与药物化学信息导航)http://wwwcmc.pharm.uu.nl/oyen/webpicks.htmlAnnals of Clinical Microbiology and Antimicrobials (免费,全文)/Annual Review of Pharmacology and Toxicology (免费,摘要)/Anti-Cancer Drug Design (免费,摘要)/antcan/生物有机化学:ScienceDirect: 在线访问Elsevier的1100种期刊全文(免费目录) (免费)/生命、环境科学综合性资源TheScientificWorld (sciBASE)/生物医药:BioMedNet: The World Wide Club for the Biological and Medical Community /BIOETHICSLINE (BIOETHICS onLINE) (免费)/BIOME (生命科学资源导航)/browse/Directory of P450-containing Systems(P450酶系目录)http://p450.abc.hu/DIRLINE (卫生与生物医药信息源库) (免费)/百名最佳生物技术网站列表(Top 100 Biotechnology WWW Sites)/top100.asp从ChemWeb检索《化学工程与生物技术文摘》库CEABA (部分免费)/课程材料:MIT生物学超文本教材 ... 7001main.html生物材料网(Biomaterials Network)/生物信息学资源导航,上海生物化学所/bio/index.htm小分子生物活性数据库ChemBank (免费)/英国剑桥医学研究委员会:分子生物学实验室LMB/biology site of the network./BIOSIS/BIOSIS/ONLINE/DBSS/biosisss.htmlCATH Protein Structure Classification (蛋白质结构分类) (免费)/bsm/cath/Databases and Tools for 3-D Protein Structure Comparison and Alignment (三维蛋白质结构对比) (免费)/ce.htmlLos Alamos National Laboratory Bioscience Division/Protein Data Bank (PDB, 蛋白质数据库) (免费)/pdb/计算分子生物学:Computational Molecular Biology at NIH/molbio/酶命名数据库(ENZYME-Enzyme nomenclature database) (免费) /enzyme/Access Excellence (有关生物、生命等的科学教育网站)/Acta Biochimica Polonica (免费,全文)http://www.actabp.pl/Acta Biotechnologica (生物技术学报) (免费,摘要)http://www.wiley-vch.d ... eticIndex/...American Institute of Biological Sciences (AIBS)/American Journal of Medical Genetics Part A (免费,摘要) http://www3.interscien ... jtoc?ID=33129Amos' WWW links page (生物大分子网络资源导航)/alinks.htmlAmersham International (英国,生物技术供应商)/。
有机化学常用期刊网址

1. ScienceDirect (SD)网址:/(1) Catalysis Communications (催化通讯)(2) Journal of Molecular Catalysis A: Chemical (分子催化A:化学)(3) Tetrahedron (T) (四面体)(4) Tetrahedron: Asymmetry (TA) (四面体:不对称)(5) Tetrahedron Letters (TL) (四面体快报)(6) Applied Catalysis A: General (应用催化A)2. EBSCOhost数据库网址:/(1) Synthetic Communcations (合成通讯)(2) Letters in Organic Chemistry (LOC)(3) Current Organic Synthesis(4) Current Organic Chemistry3. Springer数据库网址:http:// /(1) Molecules (分子)(2) Monatshefte für Ch emie / Chemical Monthly (化学月报)(3) Science in China Series B: Chemistry (中国科学B)(4) Catalysis Letts (催化快报)4. ACS Publications (美国化学会)网址:/(1) Journal of the American Chemical Society (JACS) (美国化学会志)(2) Organic Letters (OL) (有机快报)(3) The Journal of Organic Chemistry (JOC) (美国有机化学)(4) Journal of Medicinal Chemistry (JMC) (美国药物化学)(5) Chemical Reiew (化学评论)5. Royal Society of Chemistry (RSC) (英国皇家化学会)网址:/Publishing/Journals/Index.asp(1) Green Chemistry (绿色化学)(2) Chemical Communications (CC) (化学通讯)(3) Chemical Society Reviews (化学会评论)(4) Journal of the Chemical Society (化学会志)Journal of the Chemical Society, Perkin Transactions 1 (1972-2002) Journal of the Chemical Society, Perkin Transactions 2 (1972-2002) Journal of the Chemical Society B: Physical Organic (1966-1971) Journal of the Chemical Society C: Organic (1966-1971)(5) Organic & Biomolecular Chemistry (OBC) (有机生物化学)/publishing/jo ... p?type=CurrentIssue6. Wiley网址:/(1) Advanced Synthesis & Catalysis (ASC) (先进合成催化)(2) Angewandte Chemie International Edition (德国应用化学)(3) Chemistry - A European Journal (欧洲化学)(4) Chinese Journal of Chemistry (中国化学)(5) European Journal of Organic Chemistry (欧洲有机化学)(6) Helvetica Chimica Acta (瑞士化学)(7) Heteroatom Chemistry (杂原子化学)7. Ingent网址:/(1) Journal of Chemical Research (JCR) (化学研究杂志)(2) Canadian Journal of Chemistry (加拿大化学)(3) Current Organic Chemistry(4) Mini-Reviews in Organic Chemistry(5) Phosphorus, Sulfur, and Silicon and the Related Elements (磷、硫、硅和相关元素)(6) Letters in Organic Chemistry8. Taylor & Francis数据库网址:http://www.journalsonline.tandf. ... sp?referrer=default(1) Synthetic Communications(2) Journal of Sulfur Chemistry(硫化学杂志)(3) Phosphorus, Sulfur, and Silicon and the Related Elements 9. Thieme数据库网址:/(1) Synlett (合成快报)(2) Synthesis (合成)10. 日本化学会网址:(1) Chem. Lett. (CL) (化学快报)http://www.jstage.jst.go.jp/browse/cl/_vols(2) Bull. Chem. Soc. Jpn. http://www.csj.jp/journals/bcsj/index.html11. 澳大利亚化学会(Australian Journal of Chemistry)http://www.publish.csiro.au/nid/52.htm12.巴西化学会.br/13.Molecules/molecules/14.韩国化学会http://journal.kcsnet.or.kr/15.印度化学会http://www.niscair.res.in/Scienc ... hin.htm&d=test816.国际有机制备和程序(Organic Preparations and Procedures International,OPPI)/17.有机化学/index.htm有机合成:Organic Syntheses(有机合成手册), John Wiley & Sons (免费)/Named Organic Reactions Collection from the University ofOxford (有机合成中的命名反应库) (免费)/thirdyearcomputing/NamedOrganicReac...有机化学资源导航Organic Chemistry Resources Worldwide/有机合成文献综述数据库Synthesis Reviews (免费)/srev/srev.htmCAMEO (预测有机化学反应产物的软件)/products/cameo/index.shtmlCarbohydrate Letters (免费,摘要)/Carbohydrate_Letters/Carbohydrate Research (免费,摘要)/locate/carresCurrent Organic Chemistry (免费,摘要)/coc/index.htmlElectronic Encyclopedia of Reagents for Organic Synthesis (有机合成试剂百科全书e-EROS)/eros/European Journal of Organic Chemistry (免费,摘要)/jpages/1434-193X/Methods in Organic Synthesis (MOS,有机合成方法)/is/database/mosabou.htmOrganic Letters (免费,目录)/journals/orlef7/index.htmlOrganometallics (免费,目录)/journals/orgnd7/index.htmlRussian Journal of Bioorganic Chemistry (Bioorganicheskaya Khimiya) (免费,摘要)http://www.wkap.nl/journalhome.htm/1068-1620Russian Journal of Organic Chemistry (Zhurnal Organicheskoi Khimii) (免费,摘要)http://www.maik.rssi.ru/journals/orgchem.htmScience of Synthesis: Houben-Weyl Methods of Molecular Transformation /Solid-Phase Synthesis database (固相有机合成)/chem_db/sps.htmlSynthetic Communications (免费,摘要)/servlet/product/productid/SCCSyntheticPages (合成化学数据库) (免费)/The Complex Carbohydrate Research Center (复杂碳水化合物研究中心) /合成材料老化与应用 (免费,目录)/default.html金属卡宾络合物催化的烯烃复分解反应 (免费)/html/books/O61BG/b1/2002/2.6%20.htm上海化学试剂研究所/英国化学数据服务中心CDS (Chemical Database Service)/cds/cds.html英国皇家化学会碳水化合物研究组织 (Carbohydrate Group of the Royal Society of Chemistry)/lap/rsccom/dab/perk002.htm有机反应催化学会 (ORCS, Organic Reaction Catalysis Society)/有机合成练习 (免费)/中国科学院成都有机化学研究所:催化与环境工程研究发展中心/MainIndex.htm金属有机及元素有机化学:CASREACT - Chemical Reactions Database(CAS的化学反应数据库)/CASFILES/casreact.html日本丰桥大学 Jinno实验室的研究数据库(液相色谱、多环芳烃/药物/杀虫剂的紫外谱、物性) (免费)http://chrom.tutms.tut.ac.jp/JINNO/ENGLISH/RESEARCH/research...A New Framework for Porous Chemistry (金属有机骨架) (免费)/alchem/articles/1056983432324.htmlActa Crystallographica Section B (免费,摘要)/b/journalhomepage.htmlActa Crystallographica Section E (免费,摘要)/e/journalhomepage.htmlBibliographic Notebooks for Organometallic Chemistryhttp://www.ensc-lille.fr/recherche/cbco/bnoc.htmlBiological Trace Element Research (生物痕量元素研究杂志) (免费,摘要) /JournalDetail.pasp?issn=0163-4984... Journal of Organometallic Chemistry (免费,摘要)/locate/jnlabr/jomOrganic Letters (免费,目录)/journals/orlef7/index.htmlOrganometallics (免费,目录)/journals/orgnd7/index.htmlSyntheticPages (合成化学数据库) (免费)/金属卡宾络合物催化的烯烃复分解反应 (免费)/html/books/O61BG/b1/2002/2.6%20.htm金属有机参考读物:The Organometallic HyperTextBook by Rob Toreki /organomet/index.html金属有机化学国家重点实验室,中国科学院上海有机所/元素有机化学国家重点实验室(南开大学)/在线网络课程:有机金属反应和均相催化机理 (Dermot O'Hare 主讲)/icl/dermot/organomet/药物化学:Fisher Scientific/PubMed: MEDLINE和PREMEDLINE (免费)/PubMed/生物医药:BioMedNet: The World Wide Club for the Biological and Medical Community/AIDSDRUGS (艾滋病药物) (免费)/pubs/factsheets/aidsinfs.htmlautodock (分子对接软件) (免费)/pub/olson-web/doc/autodock/DIRLINE (卫生与生物医药信息源库) (免费)/HISTLINE (医药史库) (免费)/TOXNET (化合物毒性相关数据库系列) (免费)/日本药典,第14版 (免费)http://jpdb.nihs.go.jp/jp14e/index.html小分子生物活性数据库ChemBank (免费)/Ashley Abstracts Database (药物研发、市场文献摘要) (免费)/databases/ashley/search.aspBIOSIS/BIOSIS/ONLINE/DBSS/biosisss.html从检索药物交易信息库PharmaDeals (部分免费)/从ChemWeb检索有机药物用途及别名库Negwer: organic-chemical drugs and their synonyms (部分免费)/negwer/negwersearch.html美国常用药品索引库RxList (免费)/美国国家医学图书馆NLM的免费在线数据库 (免费)/hotartcl/chemtech/99/tour/internet.html制药公司目录(Pharmaceutical Companies on Virtual Library: Pharmacy Page) /company.html37℃医学网/AAPS PharmSci (免费,全文)/Abcam Ltd.有关抗体、试剂的销售,抗体的搜索)/Acta Pharmaceutica (免费,摘要)http://public.srce.hr/acphee/Advanced Drug Delivery Reviews (免费,摘要)http://www.elsevier.nl/locate/drugdelivAmerican Journal of Drug and Alcohol Abuse (免费,摘要)/servlet/product/productid/ADAAmerican Journal of Pharmaceutical Education (AJPE) (免费,全文)/Amgen Inc. (医药)/Anita's web picks (药学与药物化学信息导航)http://wwwcmc.pharm.uu.nl/oyen/webpicks.htmlAnnals of Clinical Microbiology and Antimicrobials (免费,全文)/Annual Review of Pharmacology and Toxicology (免费,摘要)/Anti-Cancer Drug Design (免费,摘要)/antcan/生物有机化学:ScienceDirect: 在线访问Elsevier的1100种期刊全文 (免费目录) (免费) /生命、环境科学综合性资源TheScientificWorld (sciBASE)/生物医药:BioMedNet: The World Wide Club for the Biological and MedicalCommunity/BIOETHICSLINE (BIOETHICS onLINE) (免费)/BIOME (生命科学资源导航)/browse/Directory of P450-containing Systems(P450酶系目录)http://p450.abc.hu/DIRLINE (卫生与生物医药信息源库) (免费)/百名最佳生物技术网站列表 (Top 100 Biotechnology WWW Sites)/top100.asp从ChemWeb检索《化学工程与生物技术文摘》库CEABA (部分免费)/课程材料:MIT生物学超文本教材:8001/esgbio/7001main.html生物材料网 (Biomaterials Network)/生物信息学资源导航,上海生物化学所/bio/index.htm小分子生物活性数据库ChemBank (免费)/英国剑桥医学研究委员会:分子生物学实验室LMB/biology site of the network./生物有机化学:ScienceDirect: 在线访问Elsevier的1100种期刊全文 (免费目录) (免费) /生命、环境科学综合性资源TheScientificWorld (sciBASE)/生物医药:BioMedNet: The World Wide Club for the Biological and Medical Community/BIOETHICSLINE (BIOETHICS onLINE) (免费)/BIOME (生命科学资源导航)/browse/Directory of P450-containing Systems(P450酶系目录)http://p450.abc.hu/DIRLINE (卫生与生物医药信息源库) (免费)/百名最佳生物技术网站列表 (Top 100 Biotechnology WWW Sites) /top100.asp从ChemWeb检索《化学工程与生物技术文摘》库CEABA (部分免费) /课程材料:MIT生物学超文本教材:8001/esgbio/7001main.html生物材料网(Biomaterials Network)/生物信息学资源导航,上海生物化学所/bio/index.htm小分子生物活性数据库ChemBank (免费)/英国剑桥医学研究委员会:分子生物学实验室LMB/biology site of the network./。
Organic Syntheses, Coll. Vol. 6, p.967 (1988); Vol. 51, p.53 (1971).

Organic Syntheses, Coll. Vol. 6, p.967 (1988); Vol. 51, p.53 (1971).AZIRIDINES FROM β-IODOCARBAMATES: 1,2,3,4-TETRAHYDRONAPHTHALENE(1,2)IMINE[1H -Naphth[1,2-b ]azirine, 1a,2,3,7b-tetrahydro-]Submitted by C. H. Heathcock 1 and A. Hassner 2.Checked by William G. Kenyon and Richard E. Benson. 1. ProcedureA 500-ml., round-bottomed flask equipped with a reflux condenser is charged with a solution of 25 g. of potassium hydroxide in 250 ml. of 95% ethanol , to which is added 16.6 g. (0.0498 mole) of methyl (trans -2-iodo-1-tetralin)carbamate (Note 1). The resulting mixture is heated under reflux on a stream bath for 2 hours, cooled, and added to 500 ml. of water. The clear, yellow solution is shaken three times with 100-ml. portions of diethyl ether . The ether layers are combined, washed three times with 125-ml. portions of water and once with 125 ml. of a saturated sodium chloride , dried over 5 g. of anhydrous potassium carbonate , and filtered. The ether is removed by distillation on a steam bath, giving the crude imine as a yellow-brown oil (Note 2). The oil is transferred to a small flask, the container is rinsed with ether , and the rinse is added to the distillation flask. The product is collected by distillation through a small Vigreux column with warm water circulating through the condenser to prevent crystallization of the product. The fraction boiling at 80–82° (0.15–0.25 mm.) is collected as a solid that forms in the receiver, yielding 4.9–5.1 g. (68–70%) of the imine, m.p. 54–56° (Note 2); the IR spectrum has a band at 3205 cm.−1 (NH) (Note 3).2. Notes1. The methylcarbamate may be prepared by the procedure in Org. Synth., Coll. Vol. 6, 795 (1988).2. The submitters state that product, m.p. 49–51°, can be obtained by direct crystallization of the oil. The oil from a run conducted on a scale twice that described above is cooled to −15° and 30 ml. of pentane is added. Upon scratching the flask, the product crystallizes, is collected by filtration, and washed with a little cold pentane , yielding 9–10 g. (62–69%), m.p. 49–51°.3. The 1H NMR spectrum (CCl 4) shows a broad singlet centered at δ 0.7 (1H) and complex multiplets at 1.1–3.05 (6H) and 6.76–7.30 (4H).3. DiscussionThe procedure reported here, that of Hassner and Heathcock,3 is more convenient than the Wenker synthesis of aziridines 4 and appears to be more general.5 It represents a simple route from olefins to aziridines (via β-iodocarbamates).3,5,6 Aziridines are also useful as intermediates in the synthesis of amino alcohols and heterocyclic systems.5,7,8,9This preparation is referenced from:z Org. Syn. Coll. Vol. 6, 795 References and Notes1.Department of Chemistry, University of California, Berkeley, California 94720.2.Present address: Department of Chemistry, State University of New York, Binghamton, NewYork 13901.3. A. Hassner and C. Heathcock, Tetrahedron, 20, 1037 (1964).4.O. E. Paris and P. E. Fanta, J. Am. Chem. Soc., 74, 3007 (1952).5. A. Hassner and C. Heathcock, J. Org. Chem., 30, 1748 (1965).6.G. Drefahl and K. Ponsold, Chem. Ber., 93, 519 (1960).7.H. W. Heine, Angew. Chem., 74, 772 (1962) [Angew. Chem. Int. Ed. Engl., 1, 528 (1962)].8. A. Hassner, M. E. Lorber, and C. Heathcock, J. Org. Chem., 32, 540 (1967).9.L. A. Paquette and D. E. Kuhla, Tetrahedron Lett., 4517 (1967).AppendixChemical Abstracts Nomenclature (Collective Index Number);(Registry Number)β-IODOCARBAMATESethanol (64-17-5)potassium carbonate (584-08-7)ether,diethyl ether (60-29-7)sodium chloride (7647-14-5)potassium hydroxide (1310-58-3)Pentane (109-66-0)methylcarbamate1,2,3,4-Tetrahydronaphthalene(1,2)imine,1H-Naphth[1,2-b]azirine, 1a,2,3,7b-tetrahydro- (1196-87-8)Methyl (trans-2-iodo-1-tetralin)carbamate (1210-13-5)Copyright © 1921-2005, Organic Syntheses, Inc. All Rights Reserved。
Organic Syntheses, Coll. Vol. 2, p.464 (1943); Vol. 13, p.84 (1933).

Organic Syntheses, Coll. Vol. 2, p.464 (1943); Vol. 13, p.84 (1933).NITROSOMETHYLURETHANE[Carbamic acid, methylnitroso-, ethyl ester]Submitted by W. W. Hartman and Ross Phillips.Checked by Louis F. Fieser and J. T. Walker.1. ProcedureTo 206 g. (2 moles) of ethyl N-methylcarbamate(p. 278) and 600 cc. of ordinary ethyl ether in a 5-l. flask is added, along with 200 g. of ice, 650 g. (9 moles) of 96 per cent sodium nitrite(Note 1) dissolved in 1 l. of cold water. The flask is provided with a stopper carrying a thermometer, a tube to lead off evolved nitric oxide, and a separatory funnel with an extension tube reaching to the bottom of the flask. A solution of 1.2 kg. (6.7 moles) of cold 35 per cent nitric acid, prepared by pouring 600 g. (426 cc.) of concentrated acid onto 600 g. of ice, is then cautiously added through the funnel in the course of one and one-half hours. The flask is given an occasional swirl to ensure some mixing, but most of the stirring is done by the evolved gases. Ice is added as required to keep the temperature below 15°. The ether layer first becomes pale red and gradually changes to a blue-green. As soon as the color has changed to green, the ether layer is separated (Note 2), washed twice with cold water, and then with cold potassium carbonate solution until carbon dioxide is no longer evolved. The solution is dried with solid potassium carbonate, and the ether is distilled from a water bath using a 1-l. flask with a 30-cm. column arranged for vacuum distillation. The vacuum is applied as soon as most of the ether has been removed, and the flask is heated gently so that the temperature of the liquid does not exceed 45–50° (Note 3) until the pressure has been reduced below 20 mm. The yield of nitrosomethylurethane boiling at 59–61/10 mm. is 200 g. (76 per cent of the theoretical amount). The density is 1.133 at 20°.2. Notes1. A large excess of sodium nitrite is required to give a satisfactory yield. This may be due to reaction according to the following equations: Nitric oxide (NO) is lost during the reaction. It is not thought advisable to use this by passing in oxygen because of the danger of an explosion, or by passing in air because of the loss of material by evaporation.2. Nitrosomethylurethane irritates the skin.3. According to the literature, nitrosomethylurethane explodes when attempts are made to distil it at normal pressure.3. DiscussionNitrosomethylurethane has been prepared by treating ethyl methylcarbamate with sodium nitrite and sulfuric acid,1 and by passing the gases generated from arsenious oxide and nitric acid into an ethereal solution of ethyl methylcarbamate.2This preparation is referenced from:z Org. Syn. Coll. Vol. 3, 119z Org. Syn. Coll. Vol. 4, 780z Org. Syn. Coll. Vol. 5, 842References and Notes1.Klobbie, Rec. trav. chim. 9, 139 (1890).2.v. Pechmann, Ber. 28, 856 (1895); Schmidt, ibid. 36, 2477 (1903); Brühl, ibid. 36, 3635 (1903).AppendixChemical Abstracts Nomenclature (Collective Index Number);(Registry Number)arsenious oxideNitric oxide (NO)potassium carbonate (584-08-7)sulfuric acid (7664-93-9)ether,ethyl ether (60-29-7)nitric acid (7697-37-2)oxygen (7782-44-7)sodium nitrite (7632-00-0)carbon dioxide (124-38-9)nitric oxideNitrosomethylurethaneEthyl N-methylcarbamate,ethyl methylcarbamate (105-40-8)Carbamic acid, methylnitroso-, ethyl ester (615-53-2)Copyright © 1921-2005, Organic Syntheses, Inc. All Rights Reserved。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
Organic Syntheses, Coll. Vol. 5, p.277 (1973); Vol. 48, p.56 (1968).CYCLODECANONESubmitted by R. D. Burpitt and J. G. Thweatt1.Checked by William G. Dauben, Michael H. McGann, and Noel Vietmeyer.1. ProcedureTo a 500-ml. round-bottomed flask fitted with a 25- to 30-cm. column packed with glass helices to which is attached a water separator2 filled with hexane(Note 1) are added 126 g. (1.00 mole) of cyclooctanone(Note 2), 100 g. (1.4 moles) of pyrrolidine, 100 ml. of xylene, and 0.5 g. of p-toluenesulfonic acid. The solution is heated under reflux until the separation of water ceases (Note 3). The water separator is replaced by a distillation head, and the reaction mixture is distilled through the column under reduced pressure to remove solvent and unreacted starting materials. When the head temperature reaches 50° (1 mm.), distillation is stopped, and the residue of almost pure N-(1-cycloocten-1-yl)pyrrolidine (152–161 g.) is used in the next step without further purification (Note 4).The crude enamine is dissolved in 450 ml. of ether, and the solution is transferred to a 1-l. three-necked flask equipped with a sealed stirrer, a 250-ml. dropping funnel, and a two-necked adapter fitted with a calcium chloride tube and a thermometer immersed in the solution. A solution of 71–76 g. (0.85–0.90 mole) (Note 5) of methyl propiolate(Caution! Methyl propiolate is a severe lachrymator and should be handled only in the hood.) in 150 ml. of ether is added dropwise. During the addition the temperature of the mixture is maintained at 25–30° by periodic cooling of the reaction flask in a dry ice-acetone bath. When the addition is almost complete, a white solid begins to separate. The mixture is stirred at 25–30° for an additional hour, cooled to 0°, and filtered to remove the solid. This is dissolved in 700 ml. of 6% hydrochloric acid(Note 6), the acidic solution is warmed at 55–60° for 1 hour, and the mixture is cooled and extracted with two 100-ml. portions of ether. The ether is removed on a steam bath, and the residue of crude methyl 10-oxocyclodec-2-ene-1-carboxylate is dissolved in 300 ml. of methanol and hydrogenated over 5 g. of 5% palladium-on-alumina catalyst at 40 p.s.i. pressure and room temperature.The catalyst is filtered, 200 g. (155 ml.) of 25% aqueous sodium hydroxide is added to the filtrate, and the mixture is heated under reflux for 1 hour. The condenser is replaced by a short Vigreux column and distillation head, and the heating is continued until most of the methanol has distilled. The two-phase residue is cooled and extracted with two 100-ml. portions of ether. The ether is removed on a steam bath, and the residue is distilled through a 20-cm. Vigreux column to yield 68–77 g. (44–50%) of cyclodecanone, b.p. 94–98° (10 mm.), m.p. 20–22° (Note 7).2. Notes1. If hexane is not used in the trap, an excessive amount of pyrrolidine is lost in the aqueous layer.2. Cyclooctanone from Aldrich Chemical Co., methyl propiolate from Farchan Research Laboratories, and pyrrolidine from Eastman Organic Chemicals were used as received.3. The reaction is usually complete after 3–6 hours at reflux. Owing to dissolved pyrrolidine, the aqueous layer amounts to 35–45 ml., and thus its volume is not a good measure of the extent of reaction.4. Pure N-(1-cycloocten-1-yl)pyrrolidine, b.p. 76–78° (1 mm.), may be isolated by distillation through a Vigreux column.5. The amount used should be adjusted to be equimolar with the amount of crude enamine.6. This solid intermediate is reasonably stable to storage under nitrogen; however, the yield in the acid hydrolysis step is better when freshly prepared material is hydrolyzed immediately.7. The same reaction sequence may be used to convert cyclododecanone to cyclotetradecanone. Preparation of the pyrrolidine enamine of cyclododecanone requires 2–3 days at reflux, and reaction of the enamine with methyl propiolate is best carried out in refluxing hexane. The enamine-propiolate reaction may also be used to convert cycloheptanone to cyclononanone. In this case the procedure must be modified to provide for partial hydrogenation of the intermediate amino ester without prior hydrolysis.3 The reduced intermediate is saponified as described in the present procedure.3. DiscussionCyclodecanone has been obtained together with other products in the pyrolysis of the thorium or yttrium salts of nonanedioic acid.4 It has also been prepared by reduction of sebacoin with zinc and hydrochloric acid,5,6 by dehydration of sebacoin followed by catalytic hydrogenation,7 by ring enlargement of cyclononanone with diazomethane8,9 and of cyclooctanone with diazomethane in the presence of a Lewis acid catalyst,9 by hydroboration of 1,2-cyclodecadiene followed by oxidation of the organoborane,10 and by the present procedure.34. Merits of the PreparationThe chief merits of this preparation are its simplicity and the high purity of the product. Although the synthesis involves several steps, each step is a simple operation, and all intermediates may be used in the subsequent steps without purification. The purity of even the crude product is high, and any impurities which may be present are readily removed by a simple distillation.The overall yield of cyclodecanone is comparable to the overall yield obtained by conversion ofdimethyl sebacate to sebacoin11 and subsequent reduction to cyclodecanone.6 In addition, the present procedure does not require the use of a high-speed stirrer, the rigorous exclusion of air, and the high dilution that are necessary in preparing sebacoin.References and Notes1.Research Laboratories, Tennessee Eastman Company, Division of Eastman Kodak Company,Kingsport, Tennessee 37662.2.S. Natelson and S. Gottfried, Org. Syntheses, Coll. Vol. 3, 381 (1955).3.K. C. Brannock, R. D. Burpitt, V. W. Goodlett, and J. G. Thweatt, J. Org. Chem., 29, 818 (1964).4.L. Ruzicka, M. Stoll, and H. Schinz, Helv. Chim. Acta, 9, 249 (1926); 11, 670 (1928).5.V. Prelog, L. Frenkiel, M. Kobelt, and P. Barman, Helv. Chim. Acta, 30, 1741 (1947).6. A. C. Cope, J. W. Barthel, and R. D. Smith, Org. Syntheses, Coll. Vol. 4, 218 (1963).7.M. Stoll, Helv. Chim. Acta, 30, 1837 (1947).8. E. P. Kohler, M. Tishler, H. Potter, and H. T. Thompson, J. Am. Chem. Soc., 61, 1057 (1939).9. E. Muller, M. Bauer, and W. Rundel, Tetrahedron Lett., No. 13, 30 (1960).10. D. Devaprabhakara and P. D. Gardner, J. Am. Chem. Soc., 85, 1458 (1963).11.N. L. Allinger, Org. Syntheses, Coll. Vol. 4, 840 (1963).AppendixChemical Abstracts Nomenclature (Collective Index Number);(Registry Number)Sebacoinpalladium-on-aluminaenamine-propiolatethorium or yttrium salts of nonanedioic acidhydrochloric acid (7647-01-0)methanol (67-56-1)ether (60-29-7)sodium hydroxide (1310-73-2)nitrogen (7727-37-9)zinc (7440-66-6)xylene (106-42-3)Diazomethane (334-88-3)pyrrolidine(123-75-1)hexane (110-54-3)Cyclodecanone (1502-06-3)cyclononanone (3350-30-9)Cycloheptanone (502-42-1)dimethyl sebacate (106-79-6)Cyclooctanone (502-49-8)cyclododecanone (830-13-7)methyl propiolate (922-67-8)N-(1-cycloocten-1-yl)pyrrolidinemethyl 10-oxocyclodec-2-ene-1-carboxylatecyclotetradecanone (3603-99-4)pyrrolidine enamine1,2-cyclodecadienep-toluenesulfonic acid (104-15-4) Copyright © 1921-2005, Organic Syntheses, Inc. All Rights Reserved。