高等有机5-有机反应机理的研究和描述
《高等有机化学—反应和机理》(Bernard Miller)笔记

●Woodward-Hoffmann规则一:4n电子的热电环化反应,如果按照顺旋方式进行是允许的;4n+2电子的热电环化反应,如果按照对旋的方式进行时允许的。
●顺旋:多烯烃的末端碳原子或环烯烃的饱和碳原子,以相同方向(同为顺时针或同为逆时针)旋转成键或断键,这种方式称为顺旋。
顺旋:多烯烃的末端碳原子或环烯烃的饱和碳原子,以不相同方向旋转成键或断键,这种方式称为对旋。
●最高已占分子轨道(HOMO)在4n+2的体系中是对称的;最低未占分子轨道(LUMO)在4n+2的体系中是反对称的。
●前线轨道理论:忽略较低的能级轨道,只考虑HOMO。
前线轨道理论能简单、形象化,但是理论上不完善,在理论上应该有更精确的处理方法。
在电环化反应中,对旋是允许的,顺旋是禁阻的。
●轨道对称性守恒:反应物中的每个轨道的对称性,在反应后对称性保持不变。
●用相关图法处理电环化反应遵循轨道对称性守恒。
●相关图法处理4n+2体系的热环化反应(对旋):以1,3,5-己三烯为例:(1)形成6个分子轨道(2)用能量最低的形成键,和的对称性相同,都是镜面对称的。
(3)是由6个原子轨道组成,键是2个原子轨道组成,故转化为时,可以想象其中有4个原子轨道的系数降低为0。
(4) 1,3,5-己三烯的,不能转化为1,3环己二烯的,因为前者的的对称性是镜面反对称,后者的的对称性是镜面对称,对称性不匹配。
故1,3,5-己三烯的事转化为1,3环己二烯的,同理1,3,5-己三烯的事转化为1,3环己二烯的(5)能量分配很合理,故反应是允许的。
用相关图法处理4n体系的热环化反应(对旋):以1,3-丁二烯为例:(1)用能量最低的形成键(2)用1,3-丁二烯的形成环丁烯的;用1,3-丁二烯的形成环丁烯的。
理由同4n+2体系,因为对称性不守恒。
(3) 1,3-丁二烯的上有2个电子,而要形成的环丁烯的电子在上。
但是1,3-丁二烯要转化为环丁烯的,如果发生这样的转化,就会形成能量很高的环丁烯的激发态。
高等有机重排反应机理

(1)C3到C8的环扩大已实现,小环产率高,因为角张力的解除提供动力,环缩小已用 于四员环和C6到C8的环,环戊基阳离子缩小成环丁基体系,由于张力增大而行不通。
(2)某些羟胺发生环扩大反应:
CH2NH2 HNO2
O
OH
(3)类似的反应如溴代环醇与格氏试剂(起碱的作用)反应消去OH中的质子,加热环扩 大。
HCl
N N NH
N2Cl +
NH2
NN
NH2
OH N2Cl +
NN OH
二 立体化学的研究
如果反应物有旋光性,可以根据重排产物是否保持其旋光性来判断历程。后面讨论的贝克曼重排, 其旋光性重排后保持不变,证明其迁移基团重排过程中未脱离原来分子,否则,生成外消旋产物。
5
三 示踪原子法
用示踪原子法的方法,可以得到一些直接有用的信息。 Claisen重排是通过示踪 原子方法对其历程进一步确证的。
RCOOH + HN3
RNCO
RNH2
硫酸是常用的催化剂,有时也用Lewis酸做催化剂。长链及有位阻的羧酸反应收率较高。
酮和叠氮酸反应是使NH“插入”到羰基和R基之间,这样把酮转变成酰胺。
R C R' + HN3 H+ O
醛的重排产物通常是腈
R C NH R' O
RCHO + HN3 H
RCN + RNHCHO
CH3
少量
+ CH3
CH3 C CH CH3
CH3
主要(稳定)
CH3 +
CH3 C CH CH3 CH3
10
直接产生重排的碳正离子本身必须稳定下来,通常是失去氢, 因此产物多为烯烃。 反应特点: (1) 如果失去质子有多种选择的话,产物由Zaitsev规则决定。
化学有机反应机理

化学有机反应机理在化学领域中,有机反应机理是研究有机化合物在反应中发生的各种化学变化的关键。
它揭示了反应的基本步骤和中间体的形成,从而有助于我们理解化学反应的本质。
在本文中,我们将深入探讨有机反应机理的重要性以及其在化学领域中的应用。
一、有机反应的基本概念有机反应是指有机化合物之间或者有机化合物与其他物质之间发生的化学变化。
这些反应可以涉及单个分子的转化,也可以涉及多个分子之间的相互作用。
有机反应机理描述了这些反应的详细步骤,包括反应物的进入、中间体的形成和最终生成物的生成。
二、反应机理的研究方法了解有机反应机理的研究方法对于理解和预测反应过程至关重要。
以下是一些常用的研究方法:1. 稳定性研究:通过研究反应物和中间体的稳定性,可以初步揭示反应的可能机理路径。
2. 碰撞理论:碰撞理论认为,反应在分子之间的碰撞下发生。
通过分析反应物分子之间的相互作用,可以推断反应的机理。
3. 动力学研究:动力学研究通过测量反应速率和活化能,可以揭示反应的机理和反应势垒。
4. 光谱学方法:光谱学方法如红外光谱、核磁共振等可以通过分析反应中物质的吸收光谱,来研究反应的机理和中间体的生成。
三、常见的有机反应机理1. 取代反应:取代反应是一种常见的有机反应,其中一个官能团被另一个官能团所取代。
常见的取代反应机理包括亲核取代和电子亲核取代。
2. 加成反应:加成反应是指两个或多个反应物结合形成一个反应物的反应。
加成反应的机理可以是电子亲和力或亲核。
3. 消除反应:消除反应是指一个分子中的两个官能团被去除,从而形成一个新的化合物。
消除反应的机理可以是酸碱催化、加热或者光照等。
4. 氧化还原反应:氧化还原反应是指有机化合物中的原子氧化态和还原态发生变化的反应。
这些反应通常涉及电子的转移。
四、有机反应机理的应用有机反应机理的研究和了解对于有机合成、药物设计和催化剂开发等领域具有重要意义。
通过深入了解反应机理,可以设计更高效、环保和高选择性的合成方法。
南开大学高等有机化学课件第四章有机反应机理的研究和描述

Ea ln k ln A RT
R: 气体常数, A: 频率因数,
在不同温度下测速率常数, 可计算出 Ea: Arrhenius活化能
Ea ΔH RT ΔS Ea log k 10 .753 log T 4.576 4.576T
4.3.1 简单速率表达式的积分形式
正常情况下动力学数据用微分方程的积分形式来处理:
如简单的一级反应和二级反应:
1 C0 一级级反 : k ln( ) t C 1 b0(a) 二级反应 : k (a0 - b0)ln t a0(b)
a, b, c: 时间t时浓度
a0, b0, c0: 起始浓度
一些反应速率方程积分形式的推导:
4.2 动力学数据(Kinetic Data)
动力学数据使我们能更详细地洞察反应机理。用跟踪反 应物消失和产物出现的方法可以测定某一个反应的速度。波 谱技术提供了一个迅速又连续地监测浓度变化的方法,因而 往往被用来测量反应进行的程度。总之,任何与一种反应物 或产物的浓度有关而且能被测量的性质,都可利用来测定反 应速度。 动力学研究的目的是为了在反应物和催化剂的浓度以及 反应速度之间建立定量关系。
k1[A][B] [C] k -1
d[D] k1 k2[C] k2 [A][B] kobs.[A][B] dt k -1
大多数反应不止一步, 可以参考一些重要的多步反应例子来得出动 力学表达式, 例如在决速步之前可以有一个快速平衡:
ROH + H+
+ ROH2
快 k1 k -1
_
ROH2 RBr +H2O
计算出一个反应的自由能变化,就使反应平衡位置的计算有了 可能,也就指出了某一化学过程的可实现性。 有兴趣的反应大多数发生在溶液中,任何这种反应的焓、熵和 自由能都与溶剂介质有关。 但是,热力学数据并不能说明是否存在一个能量上有利的潜在 反应途径,即反应速度上的情报。因此,深入了解反应机理以及 有机反应进行是中间所经各步的速度和能量要求是极为重要的。
高等有机化学各章习题及答案 (5)
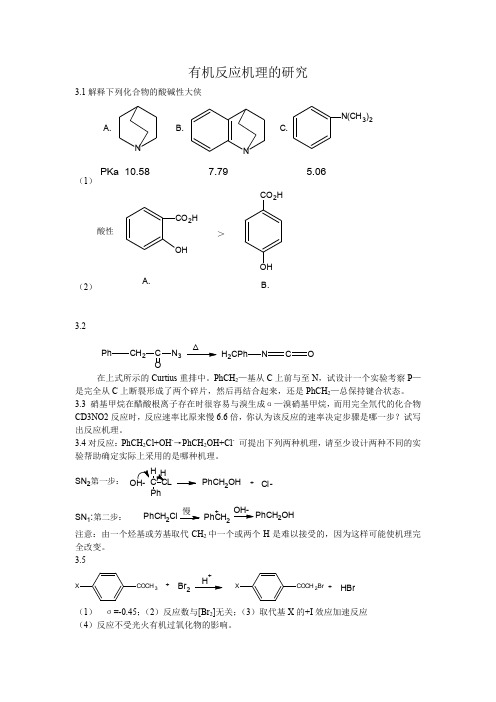
H
H
Ph C
O
C
CO
C
Me
Me
+
O
H
OH
OH
X
C
X
CH3
C +
CH3
-H+ X
C CH2
OH
O
Br-Br
X
C
X
C + HBr
CH2
CH 2Br
3.6 动力学同位素效应的转化说明反应机理发生了变化。在稀酸中,是一般酯水解机理,由
水分子进攻质子化的酯,而 D3O+的酸性比 H3O+强,质子化酯在 D2O 中比在 H2O 中浓度高, 故在 D2O 中反应速率也快。
试按以上给出的条件设计合理的反应历程。
3.6 α—醋酸—对硝基乙烯的水解是酸催化反应
OCOCH 3
O
NO2
C
H2O
NO2
C + CH3CO2H
CH2
CH3
在 6%的硫酸中,动力学同位素效应 k(H2O)/k(D2O)是 0.75,多少 69%的硫酸中,动力学
同位素效应改变为 3.25,试推测此反应的机理。
(3)SN1 机理过渡态中出现正电荷,这意味着环上的吸电子基团将使反应变慢,所以 测量环上带有 m-和 p-取代基的反应速率并计算ρ,ρ值为负时,表明是 SN1;ρ值为正则 证实为 SN2。 3.5 由给出的反应条件,可以推断这一反应为亲电反应,反应速率取决于 H+与酮络合形成碳 正离子这一步。因而推电子取代基对反应有利。碳正离子形成后迅速使 H+离解,变成取代 苯乙酮的烯醇式,由于碳碳双键上同时带了给电子的—OH 和苯环,故和 Br+的亲电加成非 常快,这样溴的浓度变化也就无关紧要了。反应可用如下历程表示:
有机化学反应机理的研究

-0.27
-0.01 -0.17 0
I
COMe CF3 NO2
+0.352
+0.376 +0.43 +0.71
+0.276
+0.502 +0.54 +0.778
由表可知,凡σ为正值的原子团都是吸引电子的原子团,而 σ为负值的原子团都是排斥电子的原子团。
2、反应常数ρ
取代基常数σ定量地表示取代基的极性效应,只 取决于取代基的结构和位置,与反应类型和反应条 件无关。 • 研究取代苯甲酸酯的水解反应,建立了关系方程 哈密特(Hammett)方程: lgK/K0 = σρ 或 lgk/k0 = σρ K和K0、k/k0分别为取代苯甲酸酯和苯甲酸酯的水 解平衡常数和速率常数 • ρ称为反应常数,是反应对取代基极性效应的敏感 性的定量尺度,ρ决定于反应特性和条件,同一类 型的反应在相同条件下ρ值相同。
五、立体化学证据 从立体化学的信息获得有关机理的证明 根据化合物的构型、构象等变化作出理性的判断
空阻差别不大, 得热力学产物
樟脑衍生物 分子中羰基 的还原
六、动力学证据
• 研究各种反应因素,如浓度、压力、温度、催化剂等对 反应速度的影响。可以提供有关反应过程的信息。 • 反应速度是反应物消失的速度或产物生成的速度 。多步 反应中,决速步骤是反应速度最慢的一步。
第二节 热力学控制及动力学控制
某一反应物A在一定条件下可能转变成两种产物B和C
速率常数 kB<kC (形成C快于B)
平衡常数 KB>KC (B比C更稳定) B
A
C
1、反应初期
⊿G C <⊿GB (活化自由能) kC>kB , A转变成C较容易, [C]>[B] 2、若平衡 主产物为C
有机化学反应机理总结

苯环亲电取代反应的一般模式
+ E+
亲电试剂
E+
-络合物
H +E
-络合物
E
+ H+
-络合物的表达方式
H
H
+H
H
E
E
E
+E
+
+
共振式
离域式
苯的硝化反应
50~60oC, 98%
+ 浓HNO3 +浓H2SO4
NO2
+ H2O
有机化合物碳上的氢被硝基取代的反应称为硝化反应
反应机理
HNO3 + H2SO4 H2O+NO2
实例:卤代烃溶剂解反应的反应机理(SN1)
进攻C+
CH3 CH3 C Br
CH3
慢 -Br-
CH3
CH3 C+
CH3
C2H5OH
快
-H+
CH3 +
CH3 C HOC2H5 CH3
CH3
CH3 C OC2H5
CH3
3 酯化反应
定义:羧酸与醇在酸的催化作用下失去一分子水 而生成酯的反应称为酯化反应
常用的催化剂有盐酸 、硫酸、苯磺酸等
实例:卤代烃双分子亲核取代反应的反应机理(SN2)
H C6H13
HO - +
Br
H3C
C6H13 H
HO
Br
CH3
C6H13 H
HO
+ Br -
CH3
有两种分子参与了决定反应速度关键步骤的亲核取代反应 称为SN2 反应
构型保持和构型翻转
n-C6H13 H C Br HOCH3
01第一章:有机化学反应机理的研究基础介绍B2

绪论一、研究有机物的过程:1、提纯结晶、蒸馏、升华、萃取、层析等2、分析元素分析、化学方法、物理方法物理方法包括:核磁共振谱、质谱、红外光谱、紫外光谱,X-ray、电子衍射等2014-5-151二、高等有机化学(Advanced Organic Chemistry)主要论述有机化合物的结构、反应机理以及它们之间的联系。
1、有机化合物的结构与性质的关系诱导效应、共轭效应、空间效应、环境效应等2、有机反应机理了解有机反应的本质,对反应结果进行解释和预测。
目前已经发现的活性中间体物种:自由基、碳负离子、碳正离子、离子游离基、卡宾、芳炔、内鎓盐等以及非碳活性中心的有机中间体。
2014-5-1522第一章:有机化学反应机理的研究基础介绍第一节:补充知识2014-5-153从下面几组数据中找找规律:(CH3)3C COOH CH3CH2COOH CH3COOH H COOH pKa 5.50 4.84 4.76 3.77I CH2COOH Br CH2COOH Cl CH2COOH F CH2COOH pKa 3.18 2.90 2.86 2.59CH3CH2CH2COOH Cl CH2CH2CH2COOH CH3CH Cl CH2COOH CH3CH2CH Cl COOH pKa 4.82 4.52 4.06 2.80+I 效应:(CH3)3C -> (CH3)2CH- > CH3CH2-> CH3--I 效应:F> Cl > Br> I2014-5-1552014-5-156诱导效应可以静电诱导方式沿着分子链由近及远地传递下去,在分子链上不会出现正负交替现象。
而且随着距离增加,诱导效应明显减弱。
一些脂肪酸和取代羧酸的pK a 值2014-5-157●饱和一元羧酸分子中,烃基上氢原子被X -、-OH 、 -NO 2等基团取代后,产生的吸电子作用,对羧基产生-I 效应,降低了羰基碳原子上的电子云密度,使得负电荷更为分散而稳定,所以酸性增强。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
如果反应速度仅与一种反应物的浓度成比例, 如果反应速度仅与一种反应物的浓度成比例, 则反应物A的浓度随时间t的变化速度为: 则反应物A的浓度随时间t的变化速度为: 反应速度= [A]/dt= 反应速度=-d[A]/dt=k[A] 服从这个速度定律的反应称为一级反应 一级反应。 服从这个速度定律的反应称为一级反应。 二级反应速度和两个反应物的浓度或一个反应 二级反应速度和两个反应物的浓度或一个反应 物浓度的平方成比例: 物浓度的平方成比例: -d[A]/dt= k[A][B] [A]/dt= [A]=[B], 若[A]=[B],则 -d[A]/dt= k[A]2 [A]/dt= 三级反应速度和三个反应物的浓度成比例 三级反应速度和三个反应物的浓度成比例 -d[A]/dt= k[A][B][C] [A]/dt= 如果[A]=[B]=[C], 如果[A]=[B]=[C],则 -d[A]/dt= k[A]3 [A]/dt=
动力学研究反应机理的局限性
反应机理的动力学上相当和 反应机理的动力学上相当 和 不能提供过 渡态的结构方面的信息。 渡态的结构方面的信息。 对于某一反应, 对于某一反应 , 有关的几个机理可以推 论出同一种速率方程, 这些机理是“ 论出同一种速率方程 , 这些机理是 “ 动 力学上相当的” 力学上相当的 ” , 所以只根据动力学是 不可能作出选择的 不可能作出选择的。
二级同位素效应:在反应中与同位素直 接相连的键不发生变化, 接相连的键不发生变化 , 而是分子中其 它化学键变化所观察到的效应, 它化学键变化所观察到的效应 , 其值通 常在K 常在KH/KD = 0.7-1.5范围内。 范围内。
过渡态
∆ GH≠
∆ GD≠
微观可逆原理
三个原子在各种距离时的位能都可由立体模 型来表示, 模型中凹处表示位能低的地方, 型来表示 , 模型中凹处表示位能低的地方 , 凸处表示位能较高的地方, 凸处表示位能较高的地方 , 这个凹凸面称为 能面。 能面。 由反应物到产物有各种可能途径, 而沿RTP 由反应物到产物有各种可能途径 , 而沿 RTP 曲线为所需能量最低, 曲线为所需能量最低 , 这就是反应将遵循的 途径。 途径。 微观可逆原理: 根据过渡态理论, 微观可逆原理 : 根据过渡态理论 , 一个反应 正向进行所经历的途径, 正向进行所经历的途径 , 也是逆向进行所要 经历的途径, 经历的途径 , 因为这个途径为这两个过程都 提供了最低的能障。 提供了最低的能障。
取代基效应
反应常数ρ值的大小, 反应常数ρ值的大小,主要反应对连在苯环上 的取代基的极性敏感性的量度, 值愈大, 的取代基的极性敏感性的量度,ρ值愈大,则 反应对极性的敏感性越高, 反应对极性的敏感性越高,并且暗示在形成过 渡态时有关的电荷要发生广泛的再分布。 渡态时有关的电荷要发生广泛的再分布。 ρ的正负也可用作判断用,如果一类反应具有 的正负也可用作判断用, 正的ρ 正的ρ值,则表示在总反应中反应中反应速度 控制步骤的过渡态中出现负电荷( 控制步骤的过渡态中出现负电荷(正电荷消 ),这些反应能被吸引电子的取代基所加速 这些反应能被吸引电子的取代基所加速; 失),这些反应能被吸引电子的取代基所加速; 如果某一类反应的ρ值为负值时, 如果某一类反应的ρ值为负值时,则表示在总 反应中反应速度控制步骤的过渡态中出现正电 荷,吸电子的取代基使这类反应的反应速度减 慢。
反应的动力学
动力学数据只提供关于决定速度步骤和在它以 动力学数据只提供关于决定速度步骤和在它以 前各步的情况, 前各步的情况,当有机反应由两步或两步以上 的基元反应组成时, 的基元反应组成时,则反应速度定律的确定一 般比较复杂,通过平衡近似和定态近似法 平衡近似和定态近似法等简 般比较复杂,通过平衡近似和定态近似法等简 化方法可求得反应的表观速度常数 反应的表观速度常数。 化方法可求得反应的表观速度常数。 动力学研究反应机理的常规顺序是:提出可能 动力学研究反应机理的常规顺序是: 的机理, 的机理,并把实验得出的速度律与根据不同可 能性推导得到的速度律作比较。 能性推导得到的速度律作比较。从而排除与观 测到的动力学不相符的机理. 测到的动力学不相符的机理.
4.1 反应的热力学
当我们研究一个有机反应时, 当我们研究一个有机反应时,最希望了解的是 这一反应将向产物方向进行到什么样程度? 这一反应将向产物方向进行到什么样程度?一 般来说, 般来说,任何体系都有转变成它们最稳定状态 的趋势,因此, 的趋势,因此,可以预料当产物的稳定性愈大 于反应物的稳定性时,则平衡愈移向产物一侧。 于反应物的稳定性时,则平衡愈移向产物一侧。 △ G=△H-T△S RTlnK 而△G=-RTlnK 要使反应发生, 要使反应发生,产物的自由能必须低于反应物 的自由能, 必须是负值。 的自由能,即△G必须是负值。
取代基效应
随着取代基与反应中心距离的加大, 随着取代基与反应中心距离的加大,其ρ 值减少,如取代羧酸在水中电离的ρ 值减少,如取代羧酸在水中电离的ρ值: ρ XC6H4CO2H 1.00 XC6H4CH2CO2H 0.49 XC6H4CH2CH2CO2H 0.21 XC6H4CH=选择苯甲酸在25℃ 哈默特选择苯甲酸在 25℃ 水溶液中的电 离作为标准反应,并指定其ρ值为1 离作为标准反应,并指定其ρ值为1,即 σ= logΚ/Κ0 因此只要测定苯甲酸和取代苯甲酸在 25℃ 水溶液中的电离常数 , 就可计算某 25℃ 水溶液中的电离常数, 一取代基的σ 一取代基的 σ 值 。 并且由式可知 , 凡 σ 为 并且由式可知, 正值的原子团都是吸引电子的原子团, 正值的原子团都是吸引电子的原子团 , 而σ为负值的原子团都是排斥电子的原子 团。
/RT
A+B
C
/RT
[A][B] = k r [A][B]
在一步反应的图中能量最高点是活化络 合物( 1a.),在它的左边, 合物(图4-1a.),在它的左边,所有络合物 都被认为同反应物处于平衡中; 都被认为同反应物处于平衡中;而在它 右边, 右边,所有络合物则被认为是同产物处 于平衡中。 于平衡中。
4.3 过渡态理论
假设一个反应先达到一个过渡态,然后 假设一个反应先达到一个过渡态, 从过渡态以极快的速度变成产物
C A+B T. S. K≠ = [T. S.] [A][B] ∆ G≠ = - RTlnK≠ κ KT 速度= [ T. S.] h [ T. S. ] = K≠ [A][B] K≠ = e-∆ G≠ κ KT e-∆ G≠ 速度= h
G
G
过渡态
∆ G1≠ C 中间体 ∆ G 2≠
∆ G 1≠
A+B 反应物
A+B 反应物 C 产物
△G
D 产物
△G
a
单步反应
b
双步 反 应
在双步反应的图b 在双步反应的图b中
反应物和产物之间包括具有一定寿命的中间体 I , 因此 ,它包含有两个过渡态 , 而且第一个过 因此,它包含有两个过渡态, 渡态的∆G 比第二个过渡态的∆G 渡态的 ∆G1≠ 比第二个过渡态的 ∆G2≠ 高 , 这意味 着第一步反应应该是速率控制步骤。 着第一步反应应该是速率控制步骤。 过渡态与中间体的区别。 过渡态与中间体的区别。中间体位于两个过渡 态之间的能量最低点,故有一定的存活期, 态之间的能量最低点,故有一定的存活期,实 际的寿命依赖于凹陷的深度。 际的寿命依赖于凹陷的深度。下凹浅暗示下一 步的活化能低,生存期短, 步的活化能低,生存期短,下凹深中间体的生 存期越长; 存期越长;而过渡态只有一个转瞬即逝的生存 并代表反应途径中的能量极大值。 期,并代表反应途径中的能量极大值。
取代基效应
脂肪族化合物和邻位取代苯衍生物, 脂肪族化合物和邻位取代苯衍生物,一 邻位取代苯衍生物 般不服从哈默特方程, 般不服从哈默特方程,由于在这些化合 物中取代基对反应的影响比较复杂, 物中取代基对反应的影响比较复杂,除 电子效应还有空间效应, 了电子效应还有空间效应,而对于各种 反应来说空间效应影响是不同的,总之, 反应来说空间效应影响是不同的,总之, 任何引起过渡态中空间情况更改的变化 也将使之不符合直线关系。 也将使之不符合直线关系。
对反应 XY + Z → YZ + X, 体系的位能总 X, 是原子相互间距离的函数, 是原子相互间距离的函数,即体系的位能 决定与它们间的相对位置及距离。
X 马鞍点 T
∆E
≠
Y
Z
P
位 能
R
Y + Z
=
Y
Z
XY + Z ∆H X + YZ 反应坐标
XY XY + Z
X+Y X + YZ 的三维位能表面图
有机反应机理的研究和描述
研究反应机理的目的 研究反应机理的目的:是认识在反应过程 目的: 中 , 发生反应体系中的原子或原子团在 结合位置、 结合位置 、 次序和结合方式上所发生的 变化,以及这种改变的方式和过程。 变化,以及这种改变的方式和过程。 反应进行的途径主要 主要由分子本身的反应 反应进行的途径主要由分子本身的反应 性能和进攻试剂的性能以及反应条件等 内外因决定。 内外因决定。
4.4 取代基效应
有机化合物的分子结构和反应性能的关系, 有机化合物的分子结构和反应性能的关系,是 高等有机化学研究的主要课题之一, 高等有机化学研究的主要课题之一,过去有不 少人对分子结构与化学活性的关系作过定量处 其中应用最广的是哈默特方程 哈默特方程。 理,其中应用最广的是哈默特方程。 哈默特指出在间和对取代苯衍生物发生反应时 在间和对取代苯衍生物发生反应时, 哈默特指出在间和对取代苯衍生物发生反应时, 取代基对反应速度或平衡常数的影响可用下式 表示: 表示: logκ/κ0 =ρlogΚ/Κ0 =σρ σ 称为 取代基常数 , 它只取决于取代基的结构 称为取代基常数 取代基常数, 和取代基的位置,与反应类型和反应条件无关, 和取代基的位置,与反应类型和反应条件无关, 它定量的表示取代基的极性效应; 称为反应 它定量的表示取代基的极性效应 ; ρ 称为 反应 常数,它决定于反应特性和条件, 常数,它决定于反应特性和条件,是反应对取 代基极性效应的敏感性的定量尺度。 代基极性效应的敏感性的定量尺度。