药材检验原始记录样本
人参原料检验记录格式
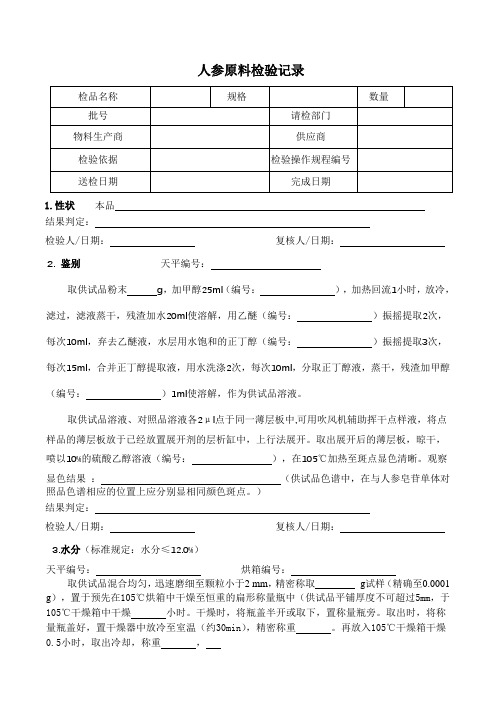
人参原料检验记录1.性状本品结果判定:检验人/日期:复核人/日期:2. 鉴别天平编号:取供试品粉末g,加甲醇25ml(编号:),加热回流1小时,放冷,滤过,滤液蒸干,残渣加水20ml使溶解,用乙醚(编号:)振摇提取2次,每次10ml,弃去乙醚液,水层用水饱和的正丁醇(编号:)振摇提取3次,每次15ml,合并正丁醇提取液,用水洗涤2次,每次10ml,分取正丁醇液,蒸干,残渣加甲醇(编号:)1ml使溶解,作为供试品溶液。
取供试品溶液、对照品溶液各2μl点于同一薄层板中,可用吹风机辅助挥干点样液,将点样品的薄层板放于已经放置展开剂的层析缸中,上行法展开。
取出展开后的薄层板,晾干,喷以10%的硫酸乙醇溶液(编号:),在105℃加热至斑点显色清晰。
观察显色结果:(供试品色谱中,在与人参皂苷单体对照品色谱相应的位置上应分别显相同颜色斑点。
)结果判定:检验人/日期:复核人/日期:3.水分(标准规定:水分≤12.0%)天平编号:烘箱编号:取供试品混合均匀,迅速磨细至颗粒小于2 mm,精密称取g试样(精确至0.0001 g),置于预先在105℃烘箱中干燥至恒重的扁形称量瓶中(供试品平铺厚度不可超过5mm,于105℃干燥箱中干燥小时。
干燥时,将瓶盖半开或取下,置称量瓶旁。
取出时,将称量瓶盖好,置干燥器中放冷至室温(约30min),精密称重。
再放入105℃干燥箱干燥0.5小时,取出冷却,称重,, , 至连续两次称重的差异不超过2mg 为止。
从减失的重量和取样量计算供试品干燥失重。
计算:试样中的水分的含量按式(1)进行计算。
100-X 3121⨯-=m m m m (1) 式中:X ——试样中水分的含量,(%);m 1 ——称量瓶和试样的质量,单位为克(g );m 2 ——称量瓶和试样干燥后的质量,单位为克(g );m 3 ——称量瓶的质量,单位为克(g )。
结果保留两位有效数字。
结果判定:检验人/日期: 复核人/日期:4.4 总灰分(标准规定:总灰分≤6.0%) 马弗炉编号:称取混合均匀的试样 g (精确至0.0001 g ),先在电炉上以小火加热使试样充分炭化至无烟,然后置于马弗炉中,在 550 ℃±25 ℃灼烧 h 。
药材检验原始记录样本

XXXXX药业(饮片)有限公司原药材检验报告单XXXXX药业(饮片)有限公司原药材检验记录【性状】结果:【鉴别】(1)显微鉴别横截面:结果:粉末:结果:(2)薄层鉴别供试品溶液的制备:取粉末1g,加乙醇15ml,加热回流1小时,放冷,滤过,滤液蒸干,残渣加乙醇5ml使溶解。
对照药材、对照品溶液配制:取菊花对照药材1g,同法制成对照药材溶液。
再取绿原酸对照品,加乙醇制成每1ml含O.5mg的溶液。
温度:(℃)相对湿度:(%)展开剂:三氯甲烷-丙酮-甲醇-5%浓氨试液(6:1:1:0.1)薄层板:硅胶G显色剂:稀碘化铋钾试液灯光:白光、紫外光灯(365nm)展距:(cm)供试品色谱中,在与对照药材色谱相对应的位置上,显相同颜色的荧光斑点。
S1为对照药材(对照品为中检所提供编号为)S2为对照品(对照品为中检所提供编号为) T为样品结果:【检查】杂质不得过 XX % (附录IX A)杂质称重: g杂质计算结果为: % (标准规定不得过 XX %)结果:膨胀度应不低于4.0(附录IX O)温度:(℃)相对湿度:(%)电子天平型号:CP214 溶剂:水样品编号 1# 2# 3#干燥品称重: g g g第一次样品膨胀后体积: ml ml ml第二次样品膨胀后体积: ml ml ml(两次差异不超过0.1ml)膨胀度计算结果为:(标准规定不低于4.0)结果:水分不得过12.0% (附录Ⅸ H 第一法)。
温度:(℃)相对湿度:(%)烘箱型号:DHG-91012SA型电子天平型号:CP214样品编号 1# 2#第一次称量瓶干燥(105℃ 3h) (g)(g)第二次称量瓶恒重(105℃ 1h) (g)(g)样品称重(g)(g)第一次称量瓶+样品干燥(105℃ 5h) (g)(g)第二次称量瓶+样品恒重(105℃ 1h) (g)(g)水分计算结果为:(%)(标准规定不得过12.0%)结果:总灰分不得过4.0%(附录Ⅸ K)温度:(℃)相对湿度:(%)马福炉型号:SX2.5-10 电子天平型号:CP214样品编号 1# 2#第一次坩锅称重(600℃ 3h) (g)(g)第二次坩锅恒重(600℃ 0.5h) (g)(g)样品称重(g)(g)第一次坩锅+残渣称重(600℃ 3h) (g)(g)第二次坩锅+残渣恒重(600℃ 0.5h) (g)(g)总灰分计算结果为:(%)(标准规定不得过4.0%)结果:酸不溶性灰分不得过3.0%(附录Ⅸ K)。
复方丹参片成品检验原始记录

复方丹参片成品检验原始记录
一、检验目的:
1.确定复方丹参片的各项质量指标是否符合药典要求;
2.检测复方丹参片中是否存在有害物质。
二、检验样品:
三、检验仪器和试剂:
1.恒温槽;
2.紫外分光光度计;
3.高效液相色谱仪;
4.显微镜;
5.乙醇;
6.氢氧化钠溶液;
7.硝酸银溶液;
8.碘液;
9.铁氯化物溶液。
四、检验项目和方法:
1.外观检查:
根据药典规定,检查复方丹参片的色泽、形状、气味等特征,判断是否符合标准。
2.含量测定:
采用高效液相色谱法测定复方丹参片中丹参酮酸B的含量。
3.汞、铅、镉、砷的含量测定:
采用草酸法测定复方丹参片中重金属的含量。
4.色谱指纹图谱分析:
采用高效液相色谱法,建立复方丹参片的色谱指纹图谱,比较样品与对照品的相似度。
5.微生物限度测试:
根据药典规定,采用菌落总数限度法和霉菌和酵母菌限度法,检测复方丹参片中的微生物限度。
五、检验结果记录:
1.外观检查:
2.含量测定:
3.汞、铅、镉、砷的含量测定:
4.色谱指纹图谱分析:
与对照品相比,复方丹参片的色谱指纹图谱相似度为98%,符合药典要求。
5.微生物限度测试:
六、检验结论:
根据上述检验结果,复方丹参片的各项质量指标均符合药典要求,未检出有害物质,微生物限度也在合理范围内,可以确认该批复方丹参片合格。
1113厚朴成品检验原始记录
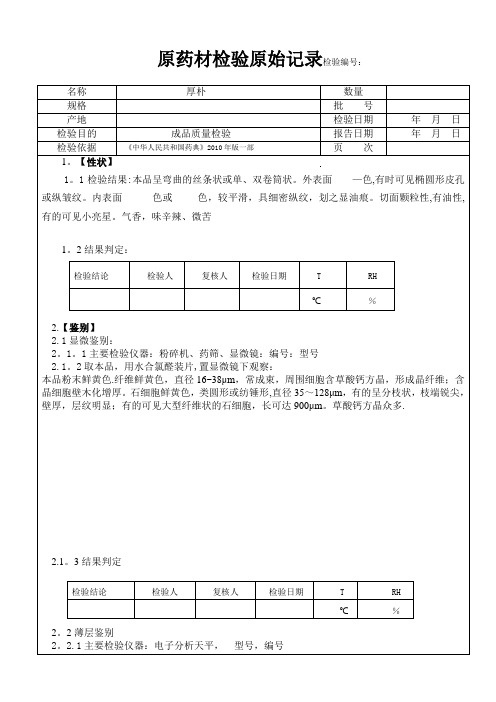
标准规定
检 验 结 果
检验结论
检验人
复核人
检验日期
供试品与对照品色谱相应的位置上,显相同颜色的斑点。
显相同颜色的
斑点□
3。水分不得过12.0%(附录ⅨH第一法).
3.1。1主要检验仪器:电子分析天平, 型号,编号
鼓风干燥箱, 型号,编号
(1)恒重称量瓶重W瓶1:
(2)恒重称量瓶重W瓶1:
7。1.4对照品溶液的制备取盐酸小檗碱对照品适量,精密称定,加流动相制成每1ml含0.1mg的溶液,即得。
7.1。5供试品溶液的制备取本品粉末(过三号筛)(样1)g(样2)g(0。1g),精密称定,置100ml量瓶中,加流动相80ml,超声处理(功率250W,频率40kHz)40分钟,放冷,用流动相稀释至刻度,摇匀,滤过,取续滤液,即得.
WX(1-水分%)X25/100
浸出物(%)1=×100%=
浸出物(%)2=×100%=
平均浸出物(%)= = =
6.3结果判定:T:℃,RH:%
项目
标准规定
检验结果
检验结论
检验人
复核人
检验时间
浸出物
≥14%
%
7【含量测定】照高效液相色谱法(附录Ⅵ D)测定。
7。1小檗碱
7.1.1主要检验仪器:电子分析天平, 型号,编号
恒重一次总重W1: 恒重一次总重W1:
恒重二次总重W2: 恒重二次总重W2:
总灰分(%)= ×100%
总灰分(%)1=×100%=
总灰分(%)2=×100%=
平均总灰分(%)= = =
4.1。2结果判定:T:℃,RH:%
项目
标准规定
药材检验原始记录样本

XXXXX药业(饮片)有限公司原药材检验报告单XXXXX药业(饮片)有限公司原药材检验记录【性状】结果:【鉴别】(1)显微鉴别横截面:结果:粉末:结果:(2)薄层鉴别供试品溶液的制备:取粉末1g,加乙醇15ml,加热回流1小时,放冷,滤过,滤液蒸干,残渣加乙醇5ml使溶解。
对照药材、对照品溶液配制:取菊花对照药材1g,同法制成对照药材溶液。
再取绿原酸对照品,加乙醇制成每1ml含O.5mg的溶液。
温度:(℃)展开剂:三氯甲烷-丙酮-甲醇-5%浓氨试液(6:1:1:0.1)薄层板:硅胶G显色剂:稀碘化铋钾试液灯光:白光、紫外光灯(365nm)展距:(cm)供试品色谱中,在与对照药材色谱相对应的位置上,显相同颜色的荧光斑点。
S1为对照药材(对照品为中检所提供编号为)S2为对照品(对照品为中检所提供编号为)T为样品结果:【检查】杂质不得过XX % (附录IX A)杂质称重: g杂质计算结果为:% (标准规定不得过XX %)结果:膨胀度应不低于4.0(附录IX O)温度:(℃)相对湿度:(%)电子天平型号:CP214 溶剂:水样品编号1# 2# 3#干燥品称重:g g g第一次样品膨胀后体积:ml ml ml第二次样品膨胀后体积:ml ml ml(两次差异不超过0.1ml)膨胀度计算结果为:(标准规定不低于4.0)结果:水分不得过12.0% (附录ⅨH 第一法)。
温度:(℃)相对湿度:(%)烘箱型号:DHG-91012SA型电子天平型号:CP214样品编号1# 2#第一次称量瓶干燥(105℃3h) (g)(g)第二次称量瓶恒重(105℃1h) (g)(g)样品称重(g)(g)第一次称量瓶+样品干燥(105℃5h) (g)(g)第二次称量瓶+样品恒重(105℃1h) (g)(g)水分计算结果为:(%)(标准规定不得过12.0%)结果:总灰分不得过4.0%(附录ⅨK)温度:(℃)相对湿度:(%)马福炉型号:SX2.5-10 电子天平型号:CP214样品编号1# 2#第一次坩锅称重(600℃3h) (g)(g)第二次坩锅恒重(600℃0.5h) (g)(g)样品称重(g)(g)第一次坩锅+残渣称重(600℃3h) (g)(g)第二次坩锅+残渣恒重(600℃0.5h) (g)(g)总灰分计算结果为:(%)(标准规定不得过4.0%)结果:酸不溶性灰分不得过3.0%(附录ⅨK)。
中药饮片——薄层扫描检验原始记录

中药饮片——薄层扫描检验原始记录XXXXXXXXX有限企业薄层扫描原始记录编号:品名规格温度编码(批号)请验日期湿度甜菜碱取本品剪碎,取约g,精细称定,加80%甲醇 50ml,加热回流 1 小时,放冷,滤过,用80%甲醇 30ml 分次清洗残渣和滤器,归并洗液与滤液,浓缩至10ml ,用盐酸调节 pH 值至 1,加人活性炭lg ,加热煮沸,放冷,滤过,用水15ml 分次清洗,归并洗液与滤液,加人新配制的 2.5%硫氰酸铬铵溶液20ml,搅匀, 100C 以下搁置 3 小时。
用G4垂熔漏斗滤过,积淀用少许冰水清洗,抽干,残渣加丙酮溶解,转移至5ml 量瓶中,加丙酮至刻度,摇匀,作为供试品溶液。
另取甜菜碱比较品适当,精细称定,加盐酸甲醇溶液(0. 5— 100)制成每 l m l 含 4 mg 的溶液,作为比较品溶液。
照薄层色谱法(公则 0502)试验。
电子天平型号:电子天平编号:薄层色谱扫描仪型号:薄层色谱扫描仪编号:(1)点样:精细汲取供试品溶液5μl、比较品溶液3μ1与6μl(2)薄层板: G 薄层板上(3)睁开剂:以丙酮 -无水乙醇 -盐酸( 10 :6 : 1)为睁开剂(4)睁开:预饱和 30 分钟,睁开,拿出,挥干溶剂,立刻喷以新配制的改进碘化铋钾试液,搁置1~3 小时至斑点清楚照(5)测试条件:波长:;λS = 515nm, λR= 590nm,丈量供试品吸光度积分值与比较品吸光度积分值 ,计算,即得。
计算:结果:(本品按干燥品计算,含甜菜碱(C5112)不得少于0. 30%)H NORSD=□切合规定□不切合规定□仅作数据累积查验人:复核人:日期:日期:。
340白芍检验原始记录-2
浙江泰康药业集团有限公司白芍检验原始记录检验内容1、【性状】:本品呈,平直或稍弯曲,两端平截,长5~18cm,直径1~2.5cm。
表面类白色或淡红棕色,光洁或有纵皱纹及细根痕,偶有残存的棕褐色外皮。
质坚实,不易折断,断面平坦,类白色或微带棕红色,形成层环明显,射线放射状。
气微、味微苦、酸。
结果:2、【鉴别】2.1、本品粉末黄白色。
糊化淀粉团块甚多。
草酸钙簇晶直径11~35µm,存在于薄壁细胞中,常排列成行,或一个细胞中含数个簇晶。
具缘纹孔及网纹导管直径20~65µm。
纤维长梭形,直径15~40µm,壁厚,微木化,具大的圆形纹孔。
结果:2.2、取本品粉末0.5g,加乙醇10ml,振摇5分钟,滤过,滤液蒸干,残渣加乙醇1ml使溶解,作为供试品溶液。
另取芍药苷对照品,加乙醇制成每1ml含1mg溶液,作为对照品溶液。
照薄层色谱法(附录VI B)试验,吸取上述两种溶液各10µl,分别点于同一硅胶G 薄层板上,以氯仿-乙酸乙酯-甲醇-甲酸(40:5:10:0.2)为展开剂,展开,取出,晾干,喷以5%香 草醛硫酸溶液,加热至斑点显色清晰。
供试品色谱中,在与 对照品色谱相应的位置上,显 的 色斑点。
结果:3、【检查】样品1:干燥的空称量瓶重M 称量瓶= 1# 2# 3# 4# gM 称量瓶+样品= g ,干燥后M ′称量瓶+样品= 1# 2# 3# 4# g 水分%=M 称量瓶+样品-M ′称量瓶+样品M 称量瓶+样品- M 称量瓶×100%=样品2:干燥的空称量瓶重M 称量瓶= 1# 2# 3# 4# gM 称量瓶+样品= g ,干燥后M ′称量瓶+样品= 1# 2# 3# 4# g 水分%=M 称量瓶+样品-M ′称量瓶+样品M 称量瓶+样品- M 称量瓶×100%=结果:3.2、总灰分:照灰分测定法依法测定。
样品1:炽灼的空坩埚重量W 1= 1# 2# 3# 4# g供试品和坩埚重量W 2= g残渣和坩埚重量W3= 1# 2# 3# 4# g总灰分%= W3-W1W2-W1×100% =样品2:炽灼的空坩埚重量W1= 1# 2# 3# 4# g供试品和坩埚重量W2= g残渣和坩埚重量W3= 1# 2# 3# 4# g总灰分%= W3-W1W2-W1×100% =结果:3.3、重金属及有害元素见后面附表。
00-002川芎检验原始记录
川芎检验原始记录检验记录编号:1 性状质量标准:本品呈不规则结节状拳形团块,直径2~7cm。
表面黄褐色,粗糙皱缩,有多数平行隆起的轮节,顶端有凹陷的类圆形茎痕,下侧及轮节上有多数小瘤状根痕,质坚实,不易折断,断面黄白色或灰黄色,散有黄棕色的油室,形成层环呈波状。
气浓香,味苦、辛,稍有麻舌感,微回甜。
结果:本项结论:□符合规定□不符合规定检验人:2 显微鉴别质量标准:□本品横切面: 木栓层为10余列细胞。
皮层狭窄,散有根迹维管束,其形成层明显。
韧皮部宽广,形成层环波状或不规则多角形。
木质部导管多角形或类圆形,大多单列或排成“V”形,偶有木纤维束。
髓部较大。
薄壁组织中散有多数油室,类圆形、椭圆形或形状不规则,淡黄棕色,靠近形成层的油室小,向外渐大;薄壁细胞中富含淀粉粒,有的薄壁细胞中含草酸钙晶体,呈类圆形团块或类簇晶状。
□本品粉末:淡黄棕色或灰棕色。
淀粉粒较多,单粒椭圆形、长圆形、类圆形、卵圆形或肾形,直径5~16μm,长约21μm,脐点点状、长缝状或人字状;偶见复粒,由2~4分粒组成。
草酸钙晶体存在于薄壁细胞中,呈类圆形团块或类簇晶状,直径10~25μm。
木栓细胞深黄棕色,表面观呈多角形,微波状弯曲。
油室多已破碎,偶可见油室碎片,分泌细胞壁薄,含有较多的油滴。
导管主为螺纹导管,亦有网纹导管及梯纹导管,直径14~50μm。
仪器及型号:□X2型双目显微镜□XSP-2C型生物显微镜结果:本项结论:□符合规定□不符合规定检验人:3 化学反应质量标准:应显红紫色取本品粉末[1g] g,加石油醚(30~60℃)5ml,放置10小时,时时振摇,静置,取上清液1ml,挥干后,残渣加甲醇1ml使溶解,再加2﹪3,5-二硝基苯甲酸的甲醇溶液2~3滴与甲醇饱和的氢氧化钾溶液2滴,观察。
本项结论:□符合规定□不符合规定检验人:4 薄层鉴别质量标准:供试品色谱中,在与川芎对照药材色谱相应的位置上,显相同颜色的荧光斑点。
仪器及型号:天平:□AB204-N电子天平□ESJ-1823电子天平□ZF—90型暗箱式紫外透射仪对照药材溶液制备:取川芎对照药材1g,同法制成对照药材溶液;对照品溶液制备:取欧当归内酯A对照品,加乙酸乙酯制成每1ml含0.1mg的溶液(置棕色量瓶中),作为对照品溶液。
吴茱萸检验原始记录
XXX药业有限公司吴茱萸检验记录【性状】本品呈形,直径mm。
表面色至褐色,粗糙,有多数点状突起或凹下的。
顶端有的裂隙,基部残留被有的果梗。
质而脆,横切面可见子房室,每室有种子1 粒。
气,味。
结论:□符合规定□不符合规定检验人:日期:年月日复核人:日期:年月日【鉴别】(1)本品粉末。
非腺毛细胞,长μm,明显,有的胞腔内含棕黄色至物。
腺毛头部细胞,,常含黄棕色内含物;柄细胞。
较多,直径μm;偶有方晶。
石细胞,直径μm,胞腔。
油室碎片有时可见,。
结论:□符合规定□不符合规定检验人:日期:年月日复核人:日期:年月日(2)薄层鉴别供试品溶液的制备:取本品粉末0.4g,加乙醇10ml,静置30分钟,超声处理30分钟,滤过。
对照品溶液配制:取吴茱萸次碱对照品(中国食品药品检定研究院批号含量),g,吴茱萸碱对照品(中国食品药品检定研究院批号含量),g,加乙醇分别制成mg/ml和mg/ml的溶液。
温度:(℃)相对湿度:(%)展开剂:石油醚(60~90℃)-乙酸乙酯-三乙胺(7:3:0.1)薄层板:硅胶G点样量:各2ul显色剂:磷钼酸硫酸溶液烘烤温度:105℃灯光:紫外光灯(365nm)展距:(cm)标准:供试品色谱中,在与对照品色谱相对应的位置上,显相同颜色的荧光斑点。
S1:吴茱萸次碱对照品溶液S2:吴茱萸碱对照品溶液T:供试品溶液检验结果:供试品色谱中,在与对照品色谱的位置上,显颜色的荧光斑点。
结论:□符合规定□不符合规定检验人:日期:年月日复核人:日期:年月日【检查】杂质不得过7%(通则2301)。
分析天平:(AUW120D)YHJ016样品重量m:g 杂质质量m杂:g ;计算公式:W(%)= m杂m× 100%检验结果:杂质为%结论:□符合规定□不符合规定检验人:日期:年月日复核人:日期:年月日水分不得过15.0%(通则0832 第二法)。
温度:(℃)相对湿度:(%)分析天平:(AUW120D)YHJ016 电热鼓风干燥箱:(101-1A)YHJ005编号1# 2#第一次称量瓶干燥(105℃ 5h) (g)(g)第二次称量瓶恒重m p(105℃ 1h) (g)(g)样品称重m y(g)(g)第一次称量瓶+样品干燥(105℃ 5h) (g)(g)第二次称量瓶+样品恒重m z(105℃ 1h) (g)(g)计算公式:水分(%)= m p+ m y - m zm y×100%检验结果:水分1# = 2# = 平均值= %结论:□符合规定□不符合规定检验人:日期:年月日复核人:日期:年月日总灰分不得过10.0%(通则2302)温度:(℃)相对湿度:(%)分析天平:(AUW120D)YHJ016 箱式电阻炉:(SX-2.5-10)YHJ006 编号1# 2# 第一次坩锅称重(600℃ 3h) (g)(g)第二次坩锅恒重m p(600℃ 0.5h) (g)(g)样品称重m y(g)(g)第一次坩锅+残渣称重(600℃ 3h) (g)(g)第二次坩锅+残渣恒重m z(600℃ 0.5h) (g)(g)计算公式:总灰分(%)= m z-m pm y×100%检验结果:总灰分1# = 2# = 平均值= %结论:□符合规定□不符合规定检验人:日期:年月日复核人:日期:年月日浸出物照醇溶性浸出物测定法(通则2201)项下的热浸法测定,用稀乙醇作溶剂,不得少于30.0%。
药品检验记录格式参考模板
%1=16.60×1.005×0.01381/0.2298×100%=100.26%
%1=21.72×1.005×0.01381/0.3006×100%=100.28%
%=100.26%+100.28%/2=100.27%=100.3%符合规定
【性状】为白色结晶性粉末,无臭,味微甜后转不适,水溶液显酸性反应。在乙
醇或乙醚中易溶,沸水中溶解,氯仿中略溶,水中微溶。符合规定
熔点:158.5~160.8℃符合规定
【鉴别】(1)本品水溶液+三氯化铁试液→紫堇色。符合规定
(2)红外图谱与对照图谱一致。符合规定
【检查】苯酚:本品0.10g制成的溶液与同法制成的对照液比较,颜色浅。符合规定
×××公司检测原始记录
No:文件编号:××××
检品名称
水杨酸
检品批号
0801022
规 格
25Kg/袋
包装形式
塑料袋
生产企业
×××药业有限公司
检品数量
160g
送检单位
威海职业学院生化系
检测日期
2008.12.10.
检验依据
《中华人民共和国药典》2005年版二部
报告日期200Leabharlann .12.15.记 录 内 容
检验人:××××××复核人:×××
炽灼残渣:坩埚1:36.2452g,36.2453g坩埚2:28.7638g,28.7637g
样品坩埚1:37.2461g样品坩埚2:29.7624g
恒重后样品坩埚1:36.2454g恒重后样品坩埚2:28.7639g
%1=(36.2454-36.2453)/(37.2461-36.2453)×100%=0.0001/1.0008×100%=0.01%
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
XXXXX药业(饮片)有限公司原药材检验报告单XXXXX药业(饮片)有限公司原药材检验记录【性状】结果:【鉴别】(1)显微鉴别横截面:结果:粉末:结果:(2)薄层鉴别供试品溶液的制备:取粉末1g,加乙醇15ml,加热回流1小时,放冷,滤过,滤液蒸干,残渣加乙醇5ml使溶解。
对照药材、对照品溶液配制:取菊花对照药材1g,同法制成对照药材溶液。
再取绿原酸对照品,加乙醇制成每1ml含O.5mg的溶液。
温度:(℃)相对湿度:(%)展开剂:三氯甲烷-丙酮-甲醇-5%浓氨试液(6:1:1:0.1)薄层板:硅胶G显色剂:稀碘化铋钾试液灯光:白光、紫外光灯(365nm)展距:(cm)供试品色谱中,在与对照药材色谱相对应的位置上,显相同颜色的荧光斑点。
S1为对照药材(对照品为中检所提供编号为)S2为对照品(对照品为中检所提供编号为) T为样品结果:【检查】杂质不得过 XX % (附录IX A)杂质称重: g杂质计算结果为: % (标准规定不得过 XX %)结果:膨胀度应不低于4.0(附录IX O)温度:(℃)相对湿度:(%)电子天平型号:CP214 溶剂:水样品编号 1# 2# 3#干燥品称重: g g g第一次样品膨胀后体积: ml ml ml第二次样品膨胀后体积: ml ml ml(两次差异不超过0.1ml)膨胀度计算结果为:(标准规定不低于4.0)结果:水分不得过12.0% (附录Ⅸ H 第一法)。
温度:(℃)相对湿度:(%)烘箱型号:DHG-91012SA型电子天平型号:CP214样品编号 1# 2#第一次称量瓶干燥(105℃ 3h) (g)(g)第二次称量瓶恒重(105℃ 1h) (g)(g)样品称重(g)(g)第一次称量瓶+样品干燥(105℃ 5h) (g)(g)第二次称量瓶+样品恒重(105℃ 1h) (g)(g)水分计算结果为:(%)(标准规定不得过12.0%)结果:总灰分不得过4.0%(附录Ⅸ K)温度:(℃)相对湿度:(%)马福炉型号:SX2.5-10 电子天平型号:CP214样品编号 1# 2#第一次坩锅称重(600℃ 3h) (g)(g)第二次坩锅恒重(600℃ 0.5h) (g)(g)样品称重(g)(g)第一次坩锅+残渣称重(600℃ 3h) (g)(g)第二次坩锅+残渣恒重(600℃ 0.5h) (g)(g)总灰分计算结果为:(%)(标准规定不得过4.0%)结果:酸不溶性灰分不得过3.0%(附录Ⅸ K)。
温度:(℃)相对湿度:(%)马福炉型号 SX2.5-10电子天平型号 CP214样品编号 1# 2#第一次坩埚+滤渣称重(600℃ 3h) (g)(g)第二次坩锅+滤渣称重(600℃ 0.5h) (g)(g)酸不溶性灰分计算结果为(标准规定不得过3.0%)结果:【浸出物】醇溶性浸出物测定法(附录X A)项下的热浸法取供试品 g,加乙醇100ml,静置1小时,回流1小时,精密滤取25ml,105℃烘3小时,置干燥器中冷却30分钟,迅速精密称重。
温度:(℃)相对湿度:(%)烘箱型号:DHG-91012SA型电子天平型号:CP214样品编号 1# 2#第一次蒸发皿称重(105℃ 3h) (g)(g)第二次蒸发皿恒重(105℃ 1h) (g)(g)样品称重(g)(g)蒸发皿+浸出物称重(105℃ 3h) (g)(g)本品浸出物计算公式:浸出物(%)=[(m2-m0)×100] / (25×m1)×100%式中:m0——蒸发皿重量(g);m1——样品重(g);m2——干燥后蒸发皿+浸出物重(g)。
1#浸出物=(%)2#浸出物=(%)浸出物平均值为:(%)(标准规定不得少于5.0%)结果:【含量测定】挥发油测定法(附录Ⅹ D)称取样品:(g)加水: ml加热时间:保持微沸5小时,至测定器中挥发油不再增加。
收集挥发油: ml挥发油含量: %(ml/g)挥发油含量=收集挥发油/称样量×100%(标准规定:挥发油不得少于1.0%(ml/g))结果:【含量测定】高效液相色谱法(附录Ⅵ D)1 仪器与测定条件天平室温度:(℃)天平室相对湿度:(%)仪器室温度:(℃)仪器室相对湿度:(%)仪器型号:岛津LC-10Avp高效液相色谱仪电子天平型号:CP214 超声仪型号:KQ3200E所用试剂:XXXXX色谱柱填充剂:十八烷基硅烷键合硅胶色谱柱长度:250 (mm)柱温:常温(℃)检测器:紫外检测器检测器波长:XCX(nm)流动相:XXXX流速: 1.0 (ml/min)2 溶液的制备2.1对照品溶液的制备取粉防己碱、防己诺林碱对照品精密称定 g、 g,加甲醇醇使溶解并稀释至刻度,摇匀,即得。
2.2供试品溶液的制备取供试品(过三号筛)约0.5g,精密称定,精密加入2%盐酸-甲醇溶液25ml,称定重量,加热回流30分钟,放冷,再称定重量,用2%盐酸-甲醇溶液补足减失的重量,摇匀,滤过,精密量取续滤液5ml,置10 ml量瓶中,加流动相至刻度,摇匀,即得。
3 测定方法精密吸取对照品溶液与供试品溶液各μl,注入液相色谱仪,测定,即得。
4 结果与计算4.1 系统适用性试验重复性:取对照品溶液连续进样针,主峰面积的RSD为%(标准规定应不大于2.0%);理论板数:(标准规定应不低于 4000 );分离度:见附页图谱,(标准规定应不低于1.5),。
4.2 响应因子计算对照品名称:粉防己碱、防己诺林碱对照品来源:中检所对照品批号:干燥条件::(g)对照品称重WR对照品稀释过程:对照品纯度:(%):(mg/ml)对照品浓度CR进样量:(μl):对照品峰面积AR平均峰面积A:R相对标准偏差RSD:(%)响应因子 F =计算公式:F=(A/ 进样量(μl))/ C RR4.3 样品测定实验编号 1# 2#称样量Wx:(g)(g)稀释倍数f:样品进样量:(μl)(μl):样品峰面积AX平均峰面积Ax:含量:(mg /g)(mg /g)含量平均值:(mg /g)相对标准偏差RSD:(%)计算公式:含量(mg /g)=(Ax/进样量)/F×f×1/Wx标准规定:本品按干燥品计算,含粉防己碱(C38H42N2O6)和防己诺林碱(C37H40N2O6)的总量不得少于1.6%。
结果:【含量测定】枸杞多糖紫外-可见分光光度法(附录V A)1 仪器与测定条件天平室温度:(℃)天平室相对湿度:(%)仪器室温度:(℃)仪器室相对湿度:(%)仪器型号:紫外可见分光光度计WFZUV-3802电子天平型号:CP2142 溶液的制备2.1 对照品溶液的制备取无水葡萄糖对照品25mg,精密称定,置250ml量瓶中,加水适量溶解,稀释至刻度,摇匀,即得(每1mI中含无水葡萄糖0.1mg)。
2.2 标准曲线的制备精密量取对照品溶液0.2ml、0.4ml、0.6ml、0.8ml、1.0ml,分别置具塞试管中,分别加水补至2.0ml,各精密加入5%苯酚溶液1m1,摇匀,迅速精密加入硫酸5ml,摇匀,放置10分钟,置40℃水浴中保温15分钟,取出,迅速冷却至室温,以相应的试剂为空白,照紫外-可见分光光度法(附录V A),在490nm的波长处测定吸光度,以吸光度为纵坐标,浓度为横坐标,绘制标准曲线。
2.3 供试品溶液的制备取本品粗粉约0.5g,精密称定,加乙醚100ml。
加热回流1小时,静置,放冷,小心弃去乙醚液,残渣置水浴上挥尽乙醚。
加入80%乙醇100ml,加热回流l小时.趁热滤过,滤渣与滤器用热80%乙醇30ml分次洗涤,滤渣连同滤纸置烧瓶中,加水150ml,加热回流2小时。
趁热滤过,用少量热水洗涤滤器,合并滤液与洗液,放冷,移至250ml 量瓶中,用水稀释至刻度,摇匀,精密量取1ml,置具塞试管中,加水1.0ml。
3 测定法照标准曲线的制备项下的方法,自“各精密加入5%苯酚溶液1m1”起,依法测定吸光,从标准曲线上读出供试品溶液中含葡萄糖的重量(mg),计算,即得。
标准规定:本品按干燥品计算,含枸杞多糖以葡萄糖(C6H12O6)计,不得少于1.8%。
结果:【含量测定】甜菜碱薄层色谱法(附录Ⅵ B薄层色谱扫描法)1 仪器与测定条件天平室温度:(℃)天平室相对湿度:(%)仪器室温度:(℃)仪器室相对湿度:(%)仪器型号:电子天平型号:CP214展开剂:丙酮-无水乙醇-盐酸(10:6:1)薄层板:硅胶G显色剂:新配制的改良碘化铋钾试液,放置1~3小时。
2.1 对照品溶液的制备供试品溶液的制备:取本品剪碎,取约2g,精密称定,加80%甲醇50ml,加热回流1小时,放冷,滤过,用80%甲醇:30m1分次洗涤残渣和滤器,合并洗液与滤液,浓缩至1Oml,用盐酸调节pH值至1,加入活性炭1g,加热煮沸,放冷,滤过,用水15m1分次洗涤,合并洗液与滤液,加入新配制的2.5%硫氰酸铬铵溶液20ml,搅匀,10℃以下放置3小时。
用G4垂熔漏斗滤过,沉淀用少量冰水洗涤,抽干,残渣加丙酮溶解,转移至5ml量瓶中,加丙酮至刻度,摇匀。
2.2 供试品溶液的制备对照品溶液配制:取甜菜碱对照品适量,精密称定,加盐酸甲醇溶液(O.5→100)制成每1m1含4mg的溶液。
3 测定法照薄层色谱法(附录Ⅵ B薄层色谱扫描法)进行扫描,波长:入s=515nm,入R=590nm,测量供试品吸光度积分值与对照品吸光度积分值,计算,即得。
标准规定:本品按干燥品计算,含甜菜碱(C5H11NO2)不得少于0.30%。
结果:【含量测定】化学滴定法1 仪器与测定条件天平室温度:(℃)天平室相对湿度:(%)电子天平型号:CP2142 测定方法取本品细粉约1g,精密称定,精密加入水1OOml,室温下浸泡4小时,时时振摇,滤过。
精密量取续滤液25ml,加水50ml,加酚酞指示液2滴,用氢氧化钠滴定液(0.1mol/L) 滴定,即得。
每1ml氢氧化钠滴定液(0.1mol/L)相当于6.404mg的枸橼酸(C6H87)。
3 结果与计算氢氧化钠滴定液的浓度:(mol/L)样品编号 1# 2#样品称重:(g)(g)消耗氢氧化钠滴定液:(ml)(ml)有机酸含量:(g)(g)百分含量:(%)(%)百分含量平均值:(%)相对标准偏差RSD:(%)计算公式:百分含量(%)=(m2×100)/ (25×m1)×100%式中:m1——样品重(g);m2——有机酸含量(g)。
标准规定:本品按干燥品计算,含有机酸以枸橼酸(C6H87)计,不得少于5.0%。
结果:结论:本品按《中华人民共和国药典》2010版一部标准检验上述项目。
结果:。