2012年FDA审评报告
医疗器械市场准入与技术要求美国FDA的审评流程解析

技术文件应按照FDA规定的格式进行 编写和整理,并通过电子方式提交至 FDA的电子审评系统。
04
美国FDA审评流程
审评流程概述
申请递交
制造商或申请人向FDA递交510(k)预市通知或PMA(上市前批准)申 请,包括技术文件、性能数据、临床数据(如适用)等。
批准后监管
对于获得批准的医疗器械,FDA将进行上市后监 管,包括定期审查、不良事件监测和合规检查等 。
申请人的权利与责任
申请人有权对FDA的决定提出异议或上诉,并需 承担确保器械持续符合FDA要求的责任。
05
案例分析:成功与失败经验分享
成功案例介绍
案例一:某创新型医 疗器械。成功因素包 括
提交了全面、严谨的 临床试验数据,证明 了产品的安全性和有 效性。
申请流程的时限因产品类别、申请资料的质量和完整性等因素而异,一般来说,Ⅰ类医疗器 械的审评时间较短,Ⅱ类和Ⅲ类医疗器械的审评时间较长。
FDA提供了一些加速审评的程序,如紧急使用授权(EUA)、突破性设备程序(BDP)等, 以加快某些急需或创新医疗器械的市场准入进程。
03
技术要求与审评标准
技术要求概述
审评标准与流程
PMA(Premarket Approval)申请:对于高风险医 疗器械,制造商需要提交PMA申请,包括详细的技 术文件、临床试验数据等,以证明产品的安全性和有 效性。审评流程更为严格,包括多轮评审、现场检查 等。
510(k)预市通知:对于中低风险医疗器械,制造商需 要提交510(k)预市通知,证明其产品与已上市且被 FDA认可的同类产品具有相同的安全性和有效性。审 评流程包括文件评审、可能的问题反馈和最终决定。
FDA批准前现场检查的政策要求及实施概述(1)

摘要鉴于美国药品市场在全球市场中的特殊地位,和美国监管法规FDA的cGMP 在全球法规监管政策中的指导性作用,学习和掌握该法规的要求是中国制药企业在实施国际化战略的必由之路。
批准前现场检查是美国药品审批的重要步骤之一,其目的是检查企业的现场GMP状态和检查现场原始数据是否真实及和申报资料一致,现场检查的意见对药品申请是否获得批准至关重要。
本文通过对美国FDA的药品申请批准前检查的政策要求的阐述,并分析总结若干国内企业在接受批准前检查过程中的经验教训,并将中美两国药品批准前检查政策进行了对比分析,希望藉此帮助国内企业更好地理解美国批准前检查政策的要求,在贯彻实施的过程中注意一些关键因素的把握,从而为企业顺利通过美国药品审批提供一些借鉴。
论文简要介绍了美国FDA药品申请批准前检查的政策发展历史和法规依据,并阐述了药品申请批准前检查的目的、范围、方式、实际执行流程、检查政策的基于体系的检查方式的特点,以及六大体系在检查过程中的关注重点等。
并通过国内三个企业通过FDA的批准前检查的案例分析,总结批准前检查流程中的关键环节和如何进行检查后的整改措施及检查成果和教训,强调在完成批准前检查后的维持GMP状态的重要性。
通过分析国际药品市场、中国药品市场、中国制药企业的现状及自身优缺点,论证国际认证特别是美国FDA认证是中国企业发展壮大的必由之路,及获得国际认证后的重要意义,鼓励国内企业坚定国际化战略思维。
鉴于国际法规政策的多样性,论文专门将中美两国的批准前政策的异同点进行了对比分析,并讨论了不同规定的优缺点,方便国内企业更好地理解美国政策。
国家新版的GMP即将推行,其宗旨和美国的cGMP要求更加接近,此对比分析对于国内企业理解和遵循新的中国法规也有积极意义。
关键词:药品申请批准前检查现行药品生产质量管理规范批准前检查政策基于体系的检查The Requirement and E nforcement of FDA’s Pre-approval Inspection(PAI)PolicyAbstractGiven the special position of American drug market in the global market, and the guiding function of American supervise regulations(FDA ‘s cGMP)in global regular policy, it is inevitable for Chinese drug companies to learn and master the requirements of this regulations in order to execute their globalization strategies. Pre-approval inspection is an important step of American drug approval, whose purpose is to ensure that the on-spot GMP and on-spot statistics of companies are true and the same as written in the application material. Therefore, the opinion from the on-spot check is vital to the approval of the application. This thesis, with an illustration of the checking policy requirements prior to FDA drug application approval in U.S.A. , through an analysis of some lessons and experience of some domestic companies during the checking, and a comparison of the checking policies between the two countries, aims to help domestic companies gain a better understanding of the American pre-approval inspection policy, and pay attention to some crucial factors during the execution process, thus to provide some guidance for the companies to get a smooth approval from the American drug approval.The thesis gives a brief introduction of the policy history and the regulation resources of American FDA pre-approval inspection, and an illustration of the purpose, the range, the method, and the actual execution procedure of the check. It lays emphasis on the features of the checking method and the checking policy, and the different focuses of the six systems.By analyzing the current situation and their own advantages and disadvantages of the international drug market, Chinese drug market, and Chinese drug companies, the author draws a conclusion that the international qualification, especial American approval is an inevitable path for Chinese companies to take for their development. Thesignificance of obtaining international 认证encourages domestic companies to become international.With an brief introduction of some necessary steps before the application check, and the sample analysis of the actual check of three domestic companies, the thesis illustrates the critical steps during the process and how to improve after the check. It emphasizes the importance to sustain the GMP state after the check by focusing on the check result, the lesson, and the staff training.Given the variety of international regulations and policies, the author gives a special comparison and analysis of the similarities and differences of the approval polices between China and U.S.A.. and discusses their merits and demerits, therefore hopes to give the domestic companies a better understanding of American policies.The thesis does not give too much pages to the methods and skills about how to cope with the check, with the hope that the companies can regard the basic requirements of GMP as their real target, and perfect and maintain their GMP system through the check. We found that the new Chinese GMP which is to be practice recently is highly similar with American’s cGMP, therefore this analysis is also helpful for Chinese pharmaceutical companies to understand and follow Chinese policy.Key Words: drug, application Pre-approval inspection cGMPChinese PAI policy System-based inspection目录第1章前言 (1)第2章美国FDA药品申请批准前GMP检查(PAI)的要求 (2)2.1 FDA的cGMP检查的目的、分类,检查范围和方式 (4)2.2 美国FDA的药品申请批准前GMP检查的执行流程 (6)2.3 美国FDA的药品申请批准前GMP检查的 (9)第3章中国企业通过FDA的cGMP检查的意义 (15)3.1全球市场情况分析 (15)3.2中国市场分析 (18)cGMP认证的意义分析 (19)第4章制药企业如何准备FDA药品申请批准前检查以及实施检查后的整改22 4.1制药企业如何准备FDA的批准前检查 (22)4.2迎接和陪同FDA的批准前检查 (23)第5章中国企业在准备和应对cGMP批准前检查过程中案例分析 (24)第6章中国企业通过FDA的cGMP现场检查后维持cGMP状态及应对批准后检查(Post- AI)的必要措施 (34)6.1维持良好的GMP状态的常规性条件 (34)6.2在职员工的cGMP培训 (37)第7章中美两国GMP批准前检查政策的异同点对比和先进性分析 (38)7.1中国GMP批准前检查政策的简要介绍 (38)7.2中美两国批准前检查政策的异同点对比和先进性分析 (43)7.3针对中国SFDA的批准前检查政策的建议 (49)结束语 (50)参考文献 (51)致谢 (53)第一章前言制药行业是一个非常特殊的行业,其产业应用的科学基础涵盖物理、化学、生物学、微生物学、医学、材料学、矿物学、机械、电子、光学、流体力学、计算机等多种学科;由于其产品的使用和人类健康息息相关,所以这也是一个被高度关注,关乎国家政治稳定性的特殊行业。
美国食品药品管理局(FDA)

美国食品药品治理局〔FDA〕〔一〕美国药政治理机构美国食品药品治理局〔FoodandDrugAdmistraton简称FDA〕,隶属于美国卫生教育福利部,负责全国药品、食品、生物制品、化装品、兽药、医疗器械以及诊断用品等的治理。
FDA下设药品局、食品局、兽药局、放射卫生局、生物制品局、医疗器械及诊断用品局和国家毒理研究中心、区域工作治理机构,即6个局〔有的刊物也称6个中心〕,一个中心和一个区域治理机构。
美国食品药品治理机构共有职工约7500人,FDA总部有1143人,其中药品局为350人。
药品局〔也称药品评价和研究中心〕负责人用药品审批工作,设有8个处和假设干科室。
1.药品治理处。
下设药品信息、信息系统设计、行政治理和预算、医学图书馆4个科室。
2.药品监督办公室。
下设有药品质量评价、药品标签监督、生产和产品质量、科研调查、法规等7个科室。
3.药品标准处。
设有常用药品评价、药品上市和广告2个科。
4.药品审评一处。
下设心血管——肾脏药、抗肿瘤药、营养药、医用造影外科和齿科药、肠胃药和凝血药5个科室。
5.药品审评二处。
下设抗感染药、代谢和内分泌药、抗病毒药3个科室。
6.流行病和生物统计处。
下设流行病及调查、生物统计2个科室。
7.研究处。
下设研究和测试、药物分析2个科室。
8.仿制药品处。
下设仿制药品、生物等效2个科室。
美国食品药品治理局设在华盛顿特区及马利兰州罗克威尔城,机构庞大,分支机构遍布全国各地。
为了加强药品质量治理,FDA将全国划分成6个大区,即太平洋区〔旧金山、西雅图、洛杉肌〕、西南区〔达拉斯、丹佛、堪萨斯〕、中西区〔芝加哥、明尼阿波利斯、底特律〕、东北区〔波士顿、纽约、布法罗〕、中大西洋区〔费城、辛辛那提、纽瓦克、巴尔的摩〕、东南区〔亚特兰大、纳什维尔、新奥尔良、奥兰多、波多利各的圣吉安〕。
每区设立一个大区所,大区所下又设假设干个地区所。
太平洋区的大区所所在地为旧金山,西南区的大区所所在地为达拉斯,中西区的大区所所在地为芝加哥,东北区的大区所所在地为波士顿,中大西洋区的大区所所在地为费城,东南区的大区所所在地为亚特兰大。
美国对高风险医疗器械的管理上市前审批——PMA

美国对高风险医疗器械的管理上市前审批——PMA一. 美国的医疗器械分类二. PMA 综述三. 需要履行PMA的情形四. 资料要求五. PMA补充和修正六. PMA审评程序七. PMA收费一. 美国的医疗器械分类美国的医疗器械分类:美国的医疗器械分类:美国的医疗器械分类:二. PMA 综述PMA 综述:PMA 综述:PMA 综述:PMA 综述:PMA 综述:PMA 综述:三. 需要履行PMA的情形需要履行PMA的情形:需要履行PMA的情形:需要履行PMA的情形:需要履行PMA的情形:四. 资料要求资料要求:资料要求:资料要求:资料要求:21CFR Part58资料要求:五. PMA补充和修正(PMA Supplements and Amendments )PMA补充和修正:PMA补充和修正:PMA补充和修正:六. PMA审评程序四个步骤:(一)形式审查和有限的技术审查(立档审查)(二)深入的科学审查、形式审查和质量体系审查(三)专家组审评(四)最终审议、归档、发通知PMA审评程序(一)形式审查和有限的技术审查(立档审查)PMA审评程序(一)形式审查和有限的技术审查(立档审查)PMA审评程序(一)形式审查和有限的技术审查(立档审查)PMA审评程序(一)形式审查和有限的技术审查(立档审查)PMA 审评程序(一)形式审查和有限的技术审查(立档审查)对不予立档的申请,申请人可以:PMA审评程序(二)深入的科学审查、形式审查和质量体系审查PMA审评程序(三)专家组审评PMA审评程序(三)专家组审评PMA审评程序(三)专家组审评PMA审评程序(三)专家组审评PMA审评程序(四)最终审议、归档、发通知PMA审评程序(四)最终审议、归档、发通知PMA审评程序(四)最终审议、归档、发通知PMA审评程序(四)最终审议、归档、发通知PMA审评程序(四)最终审议、归档、发通知PMA审评程序(四)最终审议、归档、发通知PMA审评程序(四)最终审议、归档、发通知PMA 审评程序(四)最终审议、归档、发通知PMA审评程序(四)最终审议、归档、发通知PMA审评程序(四)最终审议、归档、发通知PMA审评程序(四)最终审议、归档、发通知。
化药IND品种首轮审评结束所需时间的分析参考模板
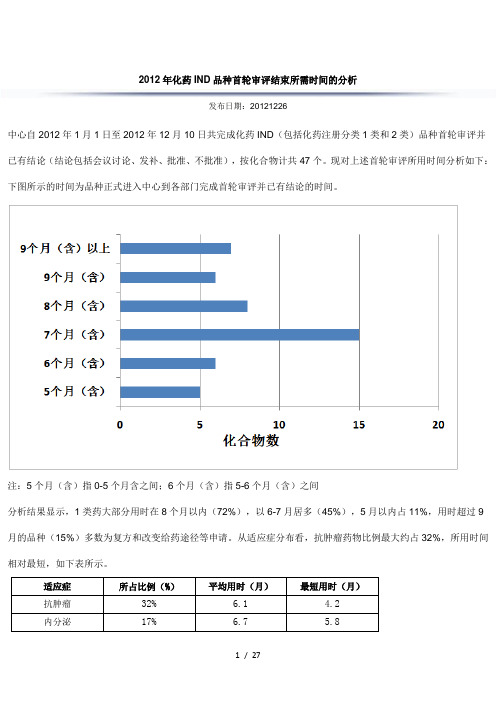
2012年化药IND品种首轮审评结束所需时间的分析发布日期:20121226中心自2012年1月1日至2012年12月10日共完成化药IND(包括化药注册分类1类和2类)品种首轮审评并已有结论(结论包括会议讨论、发补、批准、不批准),按化合物计共47个。
现对上述首轮审评所用时间分析如下:下图所示的时间为品种正式进入中心到各部门完成首轮审评并已有结论的时间。
注:5个月(含)指0-5个月含之间;6个月(含)指5-6个月(含)之间分析结果显示,1类药大部分用时在8个月以内(72%),以6-7月居多(45%),5月以内占11%,用时超过9月的品种(15%)多数为复方和改变给药途径等申请。
从适应症分布看,抗肿瘤药物比例最大约占32%,所用时间相对最短,如下表所示。
适应症所占比例(%)平均用时(月)最短用时(月)抗肿瘤32% 6.1 4.2内分泌17% 6.7 5.8对上述用时较短品种进行分析发现,具有以下特点:1、针对未被满足的临床需求,研发思路清晰。
充分掌握所涉及适应症及目标人群的特点,包括病因、流行病基础、临床预后等;了解当前临床实践现状及存在的不足,针对临床未被满足的临床需求,比如某些恶性肿瘤、耐药性乙肝、衰老相关疾病等;研究能紧密围绕立题及其临床开发计划与方案且思路清晰,针对当前需要解决的问题阶段性推进。
2、支持性数据充分,方案设计合理。
根据整体开发计划及阶段性解决的问题,提出拟进行的临床试验所探索的问题和暴露范围等开展研究,提供了充分的支持性数据,并在试验方案中反映当前的临床实践标准和非临床及同类产品暴露的风险。
总体上看,审评用时短的,通常是未发补的品种,提供的可评价证据能充分支持拟定的方案。
3、高效沟通交流,充分展示采集信息。
高效的沟通交流是现代药品注册管理体系中必需的关键手段。
CDE今年制定发布了沟通交流质量管理规范,强调进行双向预约的、基于关键阶段关键问题的、团队式的沟通交流机制,利于相互了解决策的逻辑和数据基础,提高决策的质量和效率。
FDA检查常见三种文件

FDA检查常见三种文件Form 483 #483表格This form with the eponymous number 483 is used by the executing Inspector (FDA Investigator) to document the deficiencies he found. It is issued at the end of the inspection and should be answered officially. This response is expected within 15 working days after its issuance. Only then it is guaranteed that the statement will be taken into account in a possible Warning Letter (see below). Sometime, in the case of reasonable compliance, no 483 is issued.483表格用于执行检查的检查员(FDA调查员)记录其所发现的缺陷。
在检查结束时检查员要签发该表格,受检企业则需要对其中缺陷进行正式回复。
对该表格中缺陷的回复需要在其签署后的15个工作日内提交。
只有在收到回复后,才会可能做出决定是否签发警告信(见下)。
有时,在达到合理符合的情况下,没有483表格签出来。
EIR: Establishment Investigation Report 工厂调查报告The EIR is also created by the Inspector in addition to the form 483. This should be done within 30 working days. The EIR is then examined by the responsible Center or District Office of the FDA, issuing the following statuses:除了483表以外,检查员还要制作EIR。
BCS分类 FDA 和 NICHD2012 年发布的数据
•
WATER, REFRIGERATION
•
PROBLEMS
o T echnical/Scientific o B usiness: potential population affected
• Children • Persons with swallowing problems: elderly, stroke, cerebral palsy
o Cores:
o Management o Clinical trials performance o Formulations development for clinical trials o Clinical pharmacology study design and analysis o Device development (validation)
•
DRUGS LACKING A PEDIATRIC FORMULATION
o Hydroxyurea o Isoniazid o 6-mercaptopurine, methotrexate, 6-thioguanine, isotretinoin o L-thyroxine o Clindamycin o Prednisone, prednisolone o Baclofen o Antiretrovirals o Meropenem (concentration, volume for neonates)
•
PRIORITIZATION
o Many drugs and therapeutic areas
•
CLINICAL TRIALS
o o o o o o
Lorazepam for sedation Lorazepam for status epilepticus Nitroprusside for blood pressure reduction Baclofen (oral) for spasticity (re-formulation) Lithium for mania Meropenem for severe intra-abdominal infections in neonates (volume) o Azithromycin for Ureaplasma infections o Morphine for pain in neonates
药物临床安全性评价报告
药物临床安全性评价报告撰写规范2005年2月 美国FDA发布2009年6月 药审中心组织翻译诺华制药有限公司、赛诺菲安万特制药有限公司翻译药审中心最终核准目录I. 引言 (1)II. 临床安全性审评的一般指南 (2)A. 引言 (2)B. 术语解释 (3)C. 安全性审评概述 (4)D. 安全性和有效性数据评价方法的差异 (5)E. 确定和收集安全性审评中的原始资料 (7)F. 在最开始的时候确定重大关注问题 (8)G. 稽查原始资料 (9)H. 个体病例审评/“药物相关性”的目的 (9)III. 对安全性审评报告内容的特殊指南 (11)7.1 方法与结果 (111)7.1.1 死亡 (13)7.1.2 其它严重不良事件 (17)7.1.3 退出研究和其它显著不良事件 (18)7.1.3.1 退出的总特征 (19)7.1.3.2 与退出有关的不良事件 (180)7.1.3.3 其它显著不良事件 (219)7.1.4 其它研究策略 (220)7.1.5 常见不良事件 (203)7.1.5.1 申请人在开发方案中引出(Eliciting)不良事件的方法 (203)7.1.5.2 确定合适的不良事件分类和优选术语 (213)7.1.5.3 常见不良事件的发生率 – 对不同数据库的评估 (235)7.1.5.4 常见不良事件表 (247)7.1.5.5 鉴定常见和药物有关不良事件 (257)7.1.5.6 补充分析和探索性研究 (258)7.1.6 不常见不良事件 (269)7.1.7 实验室检查结果 (30)7.1.7.1 开发项目中的实验室检查概述 (30)7.1.7.2 选择有关研究/分析法以进行实验室检测值的药物对照比较 (31)7.1.7.3 实验室数据的标准分析和探索性研究 (32)7.1.7.3.1 对集中趋势测量的重点分析 (329)7.1.7.3.2 对异常值或从正常值向异常值变化的数值的重点分析 (329)7.1.7.3.3 显著异常值和因实验室异常而退出 (33)7.1.7.4 补充分析和探索性研究 (34)7.1.7.5 特殊评估项目:肝毒性、QTc、其它 (34)7.1.8 生命体征 (35)7.1.8.1 开发项目中生命体征检查的范围 (35)7.1.8.2 用于整体药物对照比较的研究和分析方法选择 (35)7.1.8.3 生命体征数据的标准分析和探索性研究 (35)7.1.8.3.1 对集中趋势测量的重点分析 (35)7.1.8.3.2 对异常值或从正常值向异常值变化的数值的重点分析 (35)7.1.8.3.3 显著异常值和因生命体征异常而退出 (35)7.1.8.4 补充分析和探索性研究 (35)7.1.9 心电图(ECGs) (35)7.1.9.1 开发项目中ECG检查的范围,包括对临床前结果的简要综述 (36)7.1.9.2 用于整体药物对照比较的研究和分析方法选择 (36)7.1.9.3 ECG数据的标准分析和探索性研究 (36)7.1.9.3.1 对集中趋势测量的重点分析 (36)7.1.9.3.2 对异常值或从正常值向异常值变化的数值的重点分析 (36)7.1.9.3.3 显著异常值和因ECG异常而退出 (36)7.1.9.4 补充分析与探索性研究 (36)7.1.10 免疫原性 (36)7.1.11 人类致癌性 (37)7.1.12 特殊安全性研究 (37)7.1.13 戒断现象/滥用可能性 (38)7.1.14 人类生殖和怀孕数据 (38)7.1.15 对生长发育作用的评估 (38)7.1.16 过量给药经验 (39)7.1.17 上市后经验 (39)7.2 患者暴露量和安全性评估的充分性 (39)7.2.1 用于评价安全性原始临床数据来源介绍(暴露人群和暴露程度) (40)7.2.1.1 研究类型和设计 / 患者计数 (41)7.2.1.2 人口学 (41)7.2.1.3 暴露程度(剂量/周期) (42)7.2.2 用于安全性评价的二级临床数据来源介绍 (439)7.2.2.1 其它研究 (43)7.2.2.2 上市后经验 (43)7.2.2.3 文献 (43)7.2.3 总体临床经验的充分性 (44)7.2.4 特殊动物试验和/或体外试验的充分性 (44)7.2.5 常规临床检测的充分性 (44)7.2.6 代谢、清除率和相互作用检查的充分性 (45)7.2.7 对任何新药(特别是那些能够代表新药类型的药物)潜在不良反应评价的充分性;对进一步研究的建议 (46)7.2.8 数据质量和完整性的评估 (47)7.2.9 补充递交资料,包括安全性更新数据 (49)7.3 选择的不良反应总结、数据重要局限性和结论 (49)7.4 一般方法学 (51)7.4.1 汇总各研究数据以便评估和比较发生率 (51)7.4.1.1 汇总数据与单个研究数据比较 (51)7.4.1.2 结合数据 (549)7.4.2 探索预测性因素 (53)7.4.2.1 对不良事件检查结果剂量依赖性的探索 (53)7.4.2.2 探索不良事件检查结果的时间依赖性 (54)7.4.2.3 探索药物与人口学之间的相互作用 (55)7.4.2.4 探索药物与疾病之间的相互作用 (56)7.4.2.5 探索药物与药物之间的相互作用 (56)7.4.3 确定因果关系 (57)表格目录 (59)药物临床安全性评价报告撰写规范I. 引言本良好审评管理规范(GRP)指导原则拟帮助有关审评员从事临床安全性审评工作(属于NDA和BLA审评程序中的一部分)、为安全性审评报告的格式和内容提供标准统一的要求、同时确保关键内容和分析结果不会因疏忽大意而被遗漏。
FDA在检查观察项中引用“无效的OOS率”
FDA在检查观察项中引用“无效的OOS率”美国 FDA 在向印度一家制造商签发的 483 中提到无效的 OOS 率过高,这是 FDA 提出用于评估药品制造设施质量量度的三个指标之一。
这可能表明,即使行业迫使 FDA 延迟其质量量度计划,FDA 仍然可以并将通过检查过程来对其提出的指标开展性能评估。
FDA 在一份检查观察项的 483 报告中对于 2016 年 6 月 16 日对印度Lupin 公司一所设施的检查中观察到的批检验中无法解释的高差异率提出担心。
该483 中的第一个观察项关注未解释的差异,强调Lupin 两年内在某些批放行和稳定性测试中的超标(OOS)结果中 87% 无效。
相似的引述也出现在近期两封警告性中:4 月 3 日 Mylan 公司的警告性中提到其在印度 Nashik 的工厂在2016 年上半年中存在 72% 无效 OOS 率。
2 月 17 日 Wockhardt 公司 Morton Grove 子公司的警告信中提到 OOS 调查不彻底和不当的无效 OOS 结果,并要求列出有效期内产品的所有OOS、无效OOS、预期外和超趋势(OOT)的中控检测结果。
FDA 在其质量量度质量修订草案中阐述了无效的OOS 率量度,作为衡量实验室操作的稳定可靠性的指标。
无效的 OOS 率的定义为:报告期内,批次放行检验和长期稳定性检验的OOS结果中,因检验过程中的过失而被判定无效的OOS结果,除以批次放行和长期稳定性检验的全部OOS结果。
【FDA 2016版质量量度指南要求提交的量度详解 - II 2016/12/03】无效的OOS 率越高,意味着实验室操作的稳定性越差。
FDA 在483 中指出,Lupin 公司通常通过重新检测而不是通过科学评估或对制造过程的审查使初始 OOS 结果无效。
另外值得注意的是,对 Lupin 的检查是 FDA 检查员 Doan Nguyen 在 CDER 药品质量办公室工艺与设施办公室中分处主管 Zhigang Sun 的陪同下进行的。
sfda标准颁布件(2012)
sfda标准颁布件(2012)《SFDA标准颁布件(2012)》是中国食品药品监督管理总局于2012年发布的一系列标准。
这些标准对于规范和监管食品和药品行业具有重要意义。
本文将对其中的一些重要标准进行介绍,并分析其对行业发展的影响。
首先,2012年发布的《原料药通用质量控制技术指导原则》是一项重要标准。
该标准明确了原料药在质量控制方面的要求,包括原料药的检验、检测、质量控制以及申报文件的要求等。
这一标准的发布,提高了原料药的质量要求,保证了药品的安全性和有效性。
其次,《药物注册申请技术指导原则》也是一个重要的标准。
该标准规定了药物注册申请的要求和程序,明确了药物注册申请文件的内容和格式。
通过遵循该标准,可以提高药物注册审核的效率和质量,加强对药品的监管,确保药物的安全和有效性。
此外,《医疗器械分类目录(2012年版)》的发布也对医疗器械行业产生了重要影响。
该标准将医疗器械分为三类,分别是高风险、中风险和低风险类别,每类器械都对应着不同的管理要求和审批程序。
这一标准的发布,使得医疗器械行业的管理更加科学和规范,保障了人们使用医疗器械的安全性。
此外,《药品生产质量管理规范》也是一项重要的标准。
该标准规定了药品生产质量管理的要求,包括药品生产许可证的申请和管理、药品生产过程的控制和检验、药品质量记录和变更管理等。
遵循该标准,可以确保药品的质量稳定,防止药品生产中出现质量问题。
最后,《药品不良反应报告与监测管理规范》的发布也为药品安全管理提供了重要依据。
该标准规定了药品不良反应的报告和监测管理要求,明确了不良反应的定义和报告程序,要求药品生产企业和医疗机构建立不良反应的报告和监测管理制度。
这一标准的发布,有助于及时发现和报告药品不良反应,保护用药者的权益。
综上所述,《SFDA标准颁布件(2012)》是一系列重要的标准,对食品和药品行业的规范和监管起到了重要作用。
这些标准的发布,提高了原料药和药品的质量要求,加强了医疗器械行业的管理,保障了食品和药品的安全和有效性。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
CDER’S 2012 NMEs 39 novel new drugs in CY 2012:In Calendar Year 2012, FDA ’s Center for Drug Evaluation and Research (CDER) approved 39 novel new medicines, known as new molecular entities (NMEs).* This includes applications for both New Drug Applications (NDAs) and Biologics License Applications (BLAs). The blue bars in the chart to the right indicate the number of NMEs approved by CDER in each year of the past decade. CDER approved 39 NMEs in 2012, the highest total for this period. From 2003 through 2011 CDER has averaged about 24 NME approvals per year. The 2012 total is 63% higher than this previous nine year average.FDA is encouraged by this increase; however, it is too early to tell if it reflects a long-term trend toward increasing numbers of product approvals.Applications for new approvals remain steady Despite a higher number of NME approvals for the past two years, the number of applications CDER has been receiving for NMEs has not been consistently and significantly increasing. The green portion of th graph to the right indicate the number of new NDA and BLA applications for NMEs CDER has filed over the last ten years. From 2003 through 2011, CDER filed an average of about 32 applications for NMEs per year. Although all applications submitted in 2012 were not accepted for filing as of 12/31/12, CDER projects about 41 for 2012, roughly 28% higher than the 2003-2011 average of 32. Forty-one filings of new NME applications in CY 2012 would be the most this decade, another positive sign. However, the recent increase in NME filings is not enough to predict a trend toward sustained growth. FDA cannot expect a continuing upward trend for NME approvals until a sustained increase in the number of applications for NMEs submitted for approval is also demonstrated. From 2003 through 2011 CDER filed an average of about 32 applications for NMEs 39 NMEs approved in CY 2012 is the highest total approved by CDER in more than a decade In 2012 CDER approved 39 NME’s05101520253035404521362022182426213032392638412635343723# N M E A p p r o v a l s a n d A p p l i c a t i o n s Calendar Year NME Approvals NME Applications Filed 392003200420052006200720082009201020112012*Ten Year Historic Comparison Ten Year Historic Comparison *The final number of NME Applications filed in 2012 is projected, pending final validation of the data and dependent on the outcome of applications submitted in late 2012.The NMEs of 2012: see pages 14 & 15 for what these drugs are used for.AmyvidAubagio Belviq Bosulif choline C-11Cometriq Elelyso Eliquis ErivedgeFulyzaq Fycompa Gattex Iclusig Inlyta Jetrea Juxtapid KalydecoKyprolis Linzess Myrbetriq Neutroval Omontys Perjeta Picato Prepopikraxibacumab Signifor Sirturo Stendra Stivarga Stribild Surfaxin Synribo Tudorza Pressair Voraxaze Xeljanz Xtandi Zaltrap ZioptanIMPACTImpact on Public HealthThe 39 new molecular entities approved in CY 2012 represent the most approved by CDER in more than a decade, and many of these NMEs are notable for their potential positive impact and unique contributions to quality care and public health.First-in-ClassMore than half (51%) of the NMEs approved in CY 2012 (20 of 39) were identified by FDA as First-in-Class, meaning drugs which, for example, use a new and unique mechanism of action for treating a medical condition. First-in-Class is one indicator of the innovative nature of a drug and 51% First-in-Class approval rate suggests that the group of CY 2012 NMEs is a field of highly innovative new products.Particularly noteworthy First-in-Class products include Amyvid, the first brain scan imaging agent to help rule out Alzheimer’s disease as a cause of mental decline, Erivedge, the first FDA-approved drug for late-stage basal cell cancer, the most common form of skin cancer, Fulyzaq, the first drug approved for HIV-associated diarrhea, Kalydeco, the first cystic fibrosis drug to target the gene defect underlying the disease, Sirturo, the firstof a new class of drugs to treat multi-drug-resistant pulmonary tuberculosis, and Voraxaze, an important new treatment option for cancer patients to help them avoid the toxic effects of the drug methotrexate.Orphan DrugsThirteen of the 39 NMEs of CY 2012 (33%) were approved to treat rare or “orphan” diseases that affect 200,000 or fewer Americans. This is significant because patients with rare diseases often have few or no drug treatment options.The rare disease chronic myelogenous leukemia now has three new treatment options (Bosulif, Iclusig, and Synribo). Other noteworthy examples of rare diseases that now have new effective treatment options include homozygous hypercholesterolemia (Juxtapid), short bowel syndrome (Gattex), and Cushing’s disease (Signifor).BosulifCometriqElelysoGattexIclusigJuxtapidKalydecoKyprolisraxibacumabSigniforSirturoSynriboVoraxazeAmyvidAubagioBelviqcholine C-11CometriqErivedgeFulyzaqFycompaGattexJetreaJuxtapidKalydecoLinzessMyrbetriqPicatoraxibacumabSigniforSirturoSynriboVoraxazeFirst-in-ClassOrphan DrugsNotable NMEs of 2012: An exceptional year for quality In addition to the noteworthy examples of innovative First-in-Class and “Orphan” new products mentioned on page 4 and highlighted on these pages, the 39 NMEs approved in CY 2012 also include the following notable new products: Elelyso, for Gaucher disease, Eliquis, an anticoagulant to help prevent a type of blood clot known as a venous thromboembolism, Jetrea, to treat an eye condition called symptomatic vitreomacular adhesion, raxibacumab, to treat inhalational anthrax, and Stribild, a once-a-day combination pill to treat HIV-1 infection in adults who have never been treated for HIV infection. Other notable NME approvals of CY 2012 include innovative drugs to treat a variety of cancers, such as, Perjeta, to treat a specific form of late-stage breast cancer, Stivarga, to treat patients with colorectal cancer that has progressed after treatment and spread to other parts of the body and Xtandi for late-stage prostate cancer.Signifor : to treat Cushing’s disease Juxtapid : to treat homozygous hypercholestertolemia Amyvid : to help rule out Alzheimer’s disease as a cause of mental decline Erivedge : to treat late-stage basal cell cancer Voraxaze : to help avoid toxic effects of methotrexateFulyzaq: to treat HIV-associated diarrhea Sirturo : to treat multi-drug-resistant pulmonary tuberculosisBosulif, Iclusig, & Synribo: to treat chronic myelogenous leukemiaGattex : to treat short bowel syndrome Kalydeco : to treat cystic fibrosisINNOVATIONInnovative methods for expediting NMEs to market Many of the 39 NMEs of 2012 approved by CDER are notable for the regulatory methods CDER used to expedite the development and approval process. From time of submission to their approval dates, some drugs were under review for only a few months prior to approval. Particularly noteworthy examples of drugs approved rapidly are Iclusig, approved in 2.6 months, Xtandi, approved in 3.3 months, Kalydeco, 3.5 months, Erivedge, 4.7 months, and Stivarga, 5.0 months.Fast TrackFourteen of the 39 NMEs approved in 2012 (36%) were designated by CDER as Fast Track, meaning drugs with the potential to address unmet medical needs. Fast Track speeds new drug development and review; for instance, by increasing the levelof communication FDA allocates to developers and by enabling developers to use a “rolling review” process such that CDER can review portions of an application ahead of the submission of the full application.Priority ReviewSixteen of the 39 NMEs approved in 2012 (41%) were designated Priority Review, in which CDER determines the drug to potentially provide a significant advance in medical care and sets a targetto review the drug within six months instead of the standard 10 months.Accelerated ApprovalFour of the 39 NMEs approved in 2012 (10%) were approved under FDA’s Accelerated Approval program, which allows early approval of a drug for serious or life-threatening illness that offers a benefit over current treatments. This approval is based on a “surrogate endpoint” (e.g., a laboratory measure) or other clinical measure that FDA considers reasonably likely to predict clinical benefit. After this approval, the drug must undergo additional testing to confirm that benefit; this speeds the availability of the etriqElelysoFulyzaqIclusigInlytaKalydecoKyprolisAmyvidcholine C-11CometriqEliquisErivedgeFulyzaqIclusigJetreaIclusigKyprolisSirturoSynriboraxibacumabSirturoStivargaStribildSurfaxinVoraxazeXtandiKalydecoPerjetaraxibacumabSirturoStivargaVoraxazeXtandiZaltrapCombined expedited approval methodsDrugs are not limited to one expedited develoment and approval method. Inmany cases, CDER uses one or more of these tools to speed development andapproval. More than half (56%) of the 39 NMEs approved in CY 2012 (22 of39), were designated in one or more categories of Fast Track, Priority Review,and/or Accelerated Approval. Each of these designations helps expedite thespeed of the development and/or approval process and is designed to help bringimportant medications to the market as quickly as possible.Fast Track Priority Review Accelerated ApprovalInnovative Methods for Expediting NMEs to Market Innovative Methods for Expediting NMEs to MarketPREDICTABILITYPDUFA Target Dates MetUnder the Prescription Drug User Fee Act (PDUFA), sponsors are assessed user fees that provide FDA with the additional resources needed to meet performance goals.Throughout the year, CDER was able to meet or exceed PDUFA target dates for application review, agreed to with the pharmaceutical industry and approved by Congress. CDER met its PDUFA target dates for all but one (97%) of the NMEs approved in CY 2012.Amyvid Aubagio Belviq Bosulif choline C-11Cometriq Elelyso Eliquis Erivedge Fycompa Gattex Iclusig Inlyta Jetrea Juxtapid Kalydeco Kyprolis Linzess Myrbetriq Neutroval Omontys Perjeta Picato Prepopik raxibacumab Signifor Sirturo Stendra Stivarga Stribild Surfaxin Synribo Tudorza Pressair Voraxaze Xeljanz Xtandi Zaltrap Zioptan First Cycle Approval CDER approved most drugs (31 of 39) on the “first cycle” of review (79%), meaning without requests for additional information that would delay approval and lead to another cycle of review. 13 of the First Cycle Approvals are also designated as Priority Review drugs. This is particularly important because Priority Review drugs have the potential to serve as significant medical advances in health care.Approval in U.S. before Other Countries Comparing approval to other countries offers another measure of approval efficiency. Although regulatory processes differ widely between FDA and those of regulatory agencies in other countries, over three-quarters (77%) of the NMEs approved in CY 2012 (30 of 39) were approved first in the U.S. before any other country.Amyvid Aubagio Belviq Bosulif choline C-11Cometriq Elelyso Erivedge Fulyzaq Iclusig Aubagio Bosulif choline C-11Cometriq Erivedge Fulyzaq Fycompa Gattex Iclusig Inlyta Jetrea Juxtapid Kalydeco Kyprolis Linzess Myrbetriq Omontys Perjeta Picato Prepopik Signifor Sirturo Inlyta Jetrea Juxtapid Kalydeco Kyprolis Linzess Omontys Perjeta Picato raxibacumab CDER met its PDUFAtarget dates for all butone (97%) of the NMEsapproved in 2012Approval Predictability Approval Access ACCESS Sirturo Stivarga Stribild Surfaxin Synribo Tudorza Pressair Voraxaze Xeljanz Xtandi Zaltrap Stendra Stivarga Stribild Synribo Tudorza Pressair Voraxaze Xeljanz Xtandi Zaltrap First Cycle Approval First Approved in U.S.OVERVIEWThis document represents a broad overview of CDER approvals of new molecular entities (NMEs) for calendar year 2012.Although it is encouraging to see that the 39 NMEs approved in 2012 represent the highest total in more than a decade, the numbers have only substantially increased over the past two years, not long enough to establish a predictable trend. A continuing upward trend for the annual number of CDER’s NME approvals necessarily relies upona corresponding upward trend in the number of applications submitted for approval. Over the past decade, submissions for NMEs by the pharmaceutical and biotechnology industry havenot been increasing. In other words, over time, CDER can only approve a number of NMEs proportional to the number of applications for NMEs it receives.More important than the quantity of new drugs approved in 2012, is the quality of the new drugs the pharmaceutical industry has developed and the important new roles these drugs are serving toadvance medical care.Also noteworthy is the efficiency with which most of these drugs were reviewed and approved. CDER used a variety of “expedited development and approval” regulatory tools to speed these drugs to market.In all cases, while striving for efficiency of review and approval of applications for new drugs, CDER does not compromise its standards for demonstration of effectiveness and safety in the process.More important thanthe quantity of new drugs approved by CDER in CY 2012, is the quality of the new drugs and the important new roles they are serving to advance medical care.FirstinClassOrphanFastTrackPriorityReviewAcceleratedApprovalMetPDUFATargetDatesFirstCycleFirstApprovedinU.S.AmyvidAubagioBelviqBosulifcholine C-11CometriqElelysoEliquisErivedgeFulyzaqFycompaGattexIclusigInlytaJetreaJuxtapidKalydecoKyprolisLinzessMyrbetriqNeutrovalOmontysPerjetaPicatoPrepopikraxibacumabSigniforSirturoStendraStivargaStribildSurfaxinSynriboTudorza PressairVoraxazeXeljanzXtandiZaltrapZioptanDRUG DESIGNATIONSUMMARYDrugs that can treat unmet medical needsFast TrackDrugs with a target review of 6 monthsinstead of 10 monthsPriority ReviewDrugs that were approved without requestfor additional information that would delayapproval and lead to another cycle of reviewFirst CycleDrugs with a new and unique mechanism fortreating a medical conditionFirst-in-ClassDrugs approved for small populations ofpatients with rare diseasesOrphan DrugsDrugs that met the Prescription Drug UserFee Act target dates for reviewPDUFA Target DatesEarly approval based on markers that predicta reasonable benefit, with more testing toconfirm clinical benefit after approvalAccelerated ApprovalDrugs that were approved first in the U.S.before any other country worldwideFirst Approved in U.S.Summary SummaryTHE NMES OF 2012Drug Name Active Ingredient Date What it’s used forVoraxazeglucarpidase 1/17 To treat patients with toxic levels of methotrexate in their blood due to kidney failure.Picatoingenol mebutate 1/23For the topical treatment of actinic keratosis.Inlytaaxitinib 1/27To treat patients with advanced kidney cancer (renal cell carcinoma) who have not responded to another drug for this type of cancer.Erivedgevismodegib 1/30To treat adult patients with basal cell carcinoma, the most common type of skin cancer.Kalydecoivacaftor 1/31For the treatment of a rare form of cystic fibrosis (CF) in patients ages 6 years and older who have the specific G551D mutation in the Cystic Fibrosis Transmembrane Regulator (CFTR) gene.Zioptantafluprost 2/10For reducing elevated intraocular pressure in patients with open-angle glaucoma or ocular hypertension.Surfaxinlucinactant 3/6For the prevention of respiratory distress syndrome (RDS), a breathing disorder that affects premature infants.Omontys peginesatide3/27To treat anemia, a condition in which the body does not have enough healthy red blood cells, in adult dialysis patients who have chronic kidney disease (CKD).Amyvid Florbetapir F 184/6Used as a radioactive diagnostic agent for Positron Emission Tomography (PET) imaging of the brain to estimate β-amyloid neuritic plaque density in adult patients with cognitive impairment who are being evaluated for Alzheimer’s Disease (AD) and other causes of cognitive decline.Stendra avanafil4/27To treat erectile dysfunction.Elelyso taliglucerase alfa5/1For long-term enzyme replacement therapy to treat a form of Gaucher disease, a rare genetic disorder Perjeta pertuzumab6/8To treat patients with HER2-positive late-stage (metastatic) breast cancer.Belviq lorcaserin hydrochloride6/27For chronic weight management.Myrbetriq mirabegron6/28To treat adults with overactive bladder.Prepopik sodium picosulfate, magnesium oxide and citric acid 7/16To help cleanse the colon in adults preparing for colonoscopy.Kyprolis carfilzomib 7/20To treat patients with multiple myeloma who have received at least two prior therapies, including treatment with Velcade (bortezomib) and an immunomodulatory.Tudorza Pressair aclidinium bromide 7/23For the long-term maintenance treatment of bronchospasm associated with chronic obstructive pulmonary disease (COPD), including chronic bronchitis and emphysema.Zaltrap ziv-aflibercept 8/3For use in combination with a FOLFIRI (folinic acid, fluorouracil and irinotecan) chemotherapy regimen to treat adults with colorectal cancer. Stribild elvitegravir, cobicistat, emtricitabine, tenofovir disoproxil fumarate 8/27 A once-a-day combination pill to treat HIV-1 infection in adults who have never been treated for HIV infection.Neutroval tbo-filgrastim 8/29To reduce the time certain patients receiving cancer chemotherapy experience severe neutropenia, a decrease in infection-fighting white blood cells called neutrophils.Linzess linaclotide 8/30To treat chronic idiopathic constipation and to treat irritable bowel syndrome with constipation (IBS-C) in adults.Xtandi enzalutamide 8/31To treat men with late-stage (metastatic) castration-resistant prostate cancer that has spread or recurred, even with medical or surgical therapy to minimize testosterone.Bosulif bosutinib 9/4To treat chronic myelogenous leukemia (CML), a blood and bone marrow disease that usually affects older adults.Aubagio teriflunomide 9/12For the treatment of adults with relapsing forms of multiple sclerosis.Choline C 11 Injection Choline C 11 Injection 9/12 A Positron Emission Tomography (PET) imaging agent used to help detect recurrent prostate cancer.Stivarga regorafenib 9/27To treat patients with colorectal cancer that has progressed after treatment and spread to other parts of the body (metastatic).Jetrea ocriplasmin 10/17To treat an eye condition called symptomatic vitreomacular adhesion (VMA).Fycompa perampanel 10/22To treat partial onset seizures in patients with epilepsy ages 12 years and older.Synribo omacetaxine mepesuccinate 10/26To treat adults with chronic myelogenous leukemia (CML), a blood and bone marrow disease.Xeljanz tofacitinib 11/6To treat adults with moderately to severely active rheumatoid arthritis (RA) who have had an inadequate response to, or who are intolerant of, etriq cabozantinib 11/29To treat medullary thyroid cancer that has spread to other parts of the body (metastasized).Iclusig ponatinib 12/14To treat adults with chronic myeloid leukemia (CML) and Philadelphia chromosome positive acute lymphoblastic leukemia (Ph+ ALL), two rare blood and bone marrow diseases.raxibacumab raxibacumab 12/14To treat inhalational anthrax, a form of the infectious disease caused by breathing in the spores of the bacterium Bacillus anthracis.Signifor pasireotide 12/14To treat Cushing’s disease patients who cannot be helped through surgery Gattex teduglutide 12/21To treat adults with short bowel syndrome (SBS) who need additional nutrition from intravenous feeding (parenteral nutrition).Juxtapid lomitapide12/21To reduce low-density lipoprotein (LDL) cholesterol, total cholesterol, apolipoprotein B, and non-high-density lipoprotein (non-HDL) cholesterol in patients with homozygous familial hypercholesterolemia (HoFH).Sirturo bedaquiline12/28As part of combination therapy to treat adults with multi-drug resistant pulmonary tuberculosis (TB) when other alternatives are not available.Eliquis apixaban12/28To reduce the risk of stroke and dangerous blood clots (systemic embolism) in patients with atrial fibrillation that is not caused by a heart valve problem.Fulyzaqcrofelemer 12/31To treat HIV/AIDS patients whose diarrhea is not caused by an infection from a virus, bacteria, or parasite.Drug Name Active Ingredient Date What it’s used for。