有参考基因组的转录组生物信息分析模板
有参考基因组的转录组生物信息分析

一、生物信息分析流程获得原始测序序列(Sequenced Reads)后,在有相关物种参考序列或参考基因组的情况下,通过如下流程进行生物信息分析:二、项目结果说明1 原始序列数据高通量测序(如illumina HiSeq TM2000/MiSeq等测序平台)测序得到的原始图像数据文件经碱基识别(Base Calling)分析转化为原始测序序列(Sequenced Reads),我们称之为Raw Data或Raw Reads,结果以FASTQ(简称为fq)文件格式存储,其中包含测序序列(reads)的序列信息以及其对应的测序质量信息。
FASTQ格式文件中每个read由四行描述,如下:@EAS139:136:FC706VJ:2:2104:15343:197393 1:Y:18:ATCACGGCTCTTTGCCCTTCTCGTCGAAAATTGTCTCCTCATTCGAAACTTCTCTGT+@@CFFFDEHHHHFIJJJ@FHGIIIEHIIJBHHHIJJEGIIJJIGHIGHCCF其中第一行以“@”开头,随后为illumina 测序标识符(Sequence Identifiers)和描述文字(选择性部分);第二行是碱基序列;第三行以“+”开头,随后为illumina 测序标识符(选择性部分);第四行是对应序列的测序质量(Cock et al.)。
illumina 测序标识符详细信息如下:第四行中每个字符对应的ASCII值减去33,即为对应第二行碱基的测序质量值。
如果测序错误率用e表示,illumina HiSeq TM2000/MiSeq的碱基质量值用Qphred表示,则有下列关系:公式一:Qphred = -10log10(e)illumina Casava 1.8版本测序错误率与测序质量值简明对应关系如下:2 测序数据质量评估2.1 测序错误率分布检查每个碱基测序错误率是通过测序Phred数值(Phred score, Qphred)通过公式1转化得到,而Phred 数值是在碱基识别(Base Calling)过程中通过一种预测碱基判别发生错误概率模型计算得到的,对应关系如下表所显示:illumina Casava 1.8版本碱基识别与Phred分值之间的简明对应关系测序错误率与碱基质量有关,受测序仪本身、测序试剂、样品等多个因素共同影响。
转录组分析报告

转录组分析报告介绍转录组分析是研究基因组中转录过程的研究领域。
通过转录组分析,我们可以了解到在特定条件下细胞中正在转录的所有基因。
这些信息对于理解细胞功能、疾病发展以及生物技术的开发都非常重要。
本报告将介绍转录组分析的一般步骤和常用方法。
步骤一:实验设计转录组分析的第一步是设计实验。
在这个步骤中,我们需要确定要研究的样本类型、实验条件和重复次数。
合理的实验设计可以最大程度地减少误差,并提高结果的可靠性。
步骤二:RNA提取在转录组分析中,我们需要从样本中提取RNA。
RNA是细胞中转录的产物,它可以反映细胞中正在表达的基因信息。
RNA提取的质量和纯度对后续的转录组分析非常重要。
常用的提取方法包括酚氯仿法、磁珠法和硅胶膜法等。
步骤三:RNA测序RNA测序是转录组分析的核心步骤之一。
通过RNA测序,我们可以将RNA样本转化为对应的DNA序列,并确定每个基因的表达水平。
常见的RNA测序技术包括Sanger测序、二代测序和三代测序等。
二代测序技术如Illumina和Ion Torrent等已经成为转录组分析的主流技术。
步骤四:数据预处理RNA测序会产生大量的原始数据,这些数据需要进行预处理以去除噪音和提高数据质量。
数据预处理包括去除低质量的reads、去除接头序列、去除重复序列和过滤低表达基因等。
预处理后的数据可以为后续的分析提供可靠的基础。
步骤五:差异表达基因分析差异表达基因分析是转录组分析的重要环节之一。
通过比较不同条件下基因的表达水平,我们可以找到与特定条件相关的差异表达基因。
常用的差异表达基因分析方法包括DESeq、edgeR和limma等。
这些方法可以帮助我们发现与特定条件相关的生物学过程和信号通路。
步骤六:功能注释和富集分析一旦确定了差异表达基因,我们可以对这些基因进行功能注释和富集分析。
功能注释可以帮助我们了解差异表达基因的功能和参与的生物学过程。
而富集分析可以帮助我们发现差异表达基因在特定功能和通路中的富集情况。
有参考基因组的转录组生物信息分析模板

有参考基因组的转录组生物信息分析模板转录组是指一些特定生物体在特定时期和特定环境下,在其中一种特定的组织或细胞中所表达的所有基因的mRNA的总和。
转录组测序技术的发展使得我们能够全面了解基因的表达水平和差异,并帮助我们深入探索特定生物体的功能和特性。
本文将为您提供一个转录组生物信息分析的模板,以帮助研究者进行转录组数据分析。
一、质检与预处理1. 检查转录组测序数据的质量,使用FastQC等工具查看测序质量报告。
2. 根据报告,去除测序中存在的接头污染、低质量碱基,以及过短或过长的reads。
3. 使用Trimmomatic等工具进行reads修剪和过滤,保留高质量的reads。
二、比对到参考基因组2. 使用比对软件如Bowtie2、STAR等将reads比对到参考基因组上。
3. 根据比对结果生成BAM/SAM文件,并使用Samtools等工具对文件进行排序和索引。
三、基因表达量估计1. 使用HTSeq、featureCounts等软件对比对结果进行基因表达量估计,生成基因计数矩阵。
2. 将基因计数矩阵导入R或Python环境,进行表达量分析和统计。
3. 使用DESeq2、edgeR等软件对不同样本之间的差异表达基因进行筛选和统计。
四、差异表达基因分析1. 使用DESeq2、edgeR等软件进行差异表达基因分析,确定在不同条件下表达显著变化的基因。
2.使用热图、散点图、MA图等工具可视化差异表达基因的分布和表达模式。
五、注释分析1. 使用生物信息学工具如DAVID、enrichR等进行功能富集和通路分析,找出差异表达基因所涉及的生物学过程和通路。
2. 利用基因本体论(Gene Ontology)和KEGG数据库等进行差异表达基因的功能注释。
六、蛋白质互作网络分析1.将差异表达基因输入蛋白质互作数据库如STRING等,构建差异表达基因的蛋白质互作网络。
2. 使用Cytoscape等工具进行蛋白质互作网络的可视化和分析。
转录组有参考生物信息分析结题报告模版-V2.0

转录组有参考基因组生物信息分析结题报告获得原始测序序列(Sequenced Reads)后,并且其相应的基因组参考序列( Reference Genome )可以获得的情况下,可以用有参考基因组信息分析流程对数据进行详细的分析,分析流程图如下:1. 原始序列数据高通量测序(如Illunima HiSeq TM2000/ Miseq等测序平台)测序得到的原始图像数据文件经碱基识别(Base Calling)分析转化为原始测序序列(Sequenced Reads),我们称之为Raw Data或Raw Reads,结果以FASTQ(简称为fq)文件格式存储,其中包含测序序列(reads)的序列信息以及其对应的测序质量信息。
测序样品中真实数据随机截取结果如下:@HWI-ST1106:227:D14F6ACXX:1:1101:1202:2188 1:N:0:GCCAAT CGGATGATCTTCTTAATCTCTCCTTGCATAGTTATGAAACAGTCCGTGGACTTGCTGGAAAATCTCTCTTGAAGATGATGAAGAGATGGCCCTCTACAAT +CCCFFFDFFHHHHJJJJJIJIGGGIGICIGIIJEIIJIIJJI@DHEDHECFGGAHGGJGHIICGEEIEHGGGIECEEHH@HE>C@EBBE@CCDDCCCDDC @HWI-ST1106:227:D14F6ACXX:1:1101:1237:2217 1:N:0:GCCAAT GAAGGTGAGTCTGAGGAGGCCAAGGAGGGAATGTTTGTGAAAGGATATGTCTACTAAGATATTAGAAAGTATGTACTACTACTACTACTACATGTTTTCA +@@@FDADDFDHFHIIIDHIIJJJGICGGGCGHGFIGHBHEHHGI;BDHHCFGCHIIIIEHGIGHHIJJE7??ACHCDFFFFFEEECCEE>C>ACCCDC>@ @HWI-ST1106:227:D14F6ACXX:1:1101:1382:2195 1:N:0:GCCAAT TTTTGCAACAATGGCTTCCACCATGATGACTACTCTACCACAGTTCAATGGACTCAAACCCCAACCTTTCTCAGCTTCTCCAATTCAAGGCTTGGTGGCA +@@@DD3DDFFFF:CDGI@GIEEDH<F49C?EGFBF9?FF?C@BFEFGIII3BDDFFIIG7FFFIIBEFFIFDC3ACBDDDBD@>@AAD;;;@@####### @HWI-ST1106:227:D14F6ACXX:1:1101:1255:2239 1:N:0:GCCAAT CGGATTTTCAAGGGCCGCCGGGAGCGCACCGGACACCACGCGACGTGCGGTGCTCTTCCAGCCGCTGGACCCTACCTCCGGCTGAGCCGATTCCAGGGTG +CCCDFFFFHHH?FHIIIJJJJJIGBEHHJJBHBDDCDAC??@@BDBBBBD8BDDCDDACC@A?@BBB@<<CB?CB<AD?9<B@>(8>?395?4:(:<@## @HWI-ST1106:227:D14F6ACXX:1:1101:1423:2239 1:N:0:GCCAAT CTTGTATTGCTCTCCCACAACCCCGTTTTCACGGTTTAGGCTGCTCCCATTTCGCTCGCCGCTACTACGGGAATCGCTTTTGCTTTCTTTTCCTCTGGCT +CCCFDFFFHHHHHJJIJJJJJIJJGGIHIIGIIJGIGGIJJGGGJGIJ>FGIIGHGGBEHBCCBBDDD@BB@@<AABDDBCACDCDACDCD@:>@C::@C2.测序数据质量评估2.1 测序错误率分布检查测序错误率与碱基质量有关,受测序仪本身、测序试剂、样品等多个因素共同影响。
植物基因组与转录组的分析

植物基因组与转录组的分析近年来,随着生物信息学和计算机科学的不断发展,对植物基因组和转录组的研究也越来越深入。
基因组是组成生物体的所有基因序列的集合,而转录组则是指一个物种所有被转录为RNA的基因集合。
对植物基因组和转录组的深入研究不仅可以帮助我们更加理解植物的生长发育和适应环境能力,还可以为植物育种和改良提供科学依据。
一、植物基因组的测序和注释测序是分析植物基因组的第一步,它可以帮助我们确定基因组的大小和基因的位置。
目前,测序技术主要包括第一代测序技术和第二代测序技术。
第一代测序技术包括Sanger测序技术和454测序技术,虽然精度较高,但是效率低下,成本较高。
而第二代测序技术,如Illumina、Ion Torrent、PacBio以及Oxford Nanopore 等,由于其高通量、高精度和低成本等优点,已经成为当前主流的测序技术。
在基因组测序完成后,需要对其进行注释。
基因组注释是指确定基因的具体序列和位置等信息,也包括预测调控元件、非编码RNA和之间的相互作用等方面的信息。
基因组注释的方法主要包括题目比对、转录组测序和结构预测等。
我们需要将不同来源的数据结合起来进行注释,以最大限度地减少错误预测和漏预测的概率。
二、植物转录组的分析一旦获得了植物基因组的序列信息,我们接着需要了解基因组是如何表达为蛋白质的。
转录组就是表达的基因的RNA序列的总和。
分析植物转录组可以帮助我们更深入地研究基因表达调控机制以及物种的适应性和进化。
对植物转录组的分析主要包括差异表达基因分析、异构体分析、基因共表达网络分析和功能注释等。
其中差异表达基因分析是最为常见的方法,它通过比较不同条件下基因的表达情况,筛选出在不同条件下表达量有显著变化的基因。
三、植物基因组和转录组的应用对植物基因组和转录组的深入研究可以在植物育种和改良方面提供科学依据。
以水稻为例,基因组和转录组的研究揭示了水稻中关键基因的功能和表达调控机制,可以帮助我们更好地理解水稻的生长发育和适应环境的能力,也为水稻的育种和改良提供了新思路。
有参考基因组_转录组ref流程工作手册
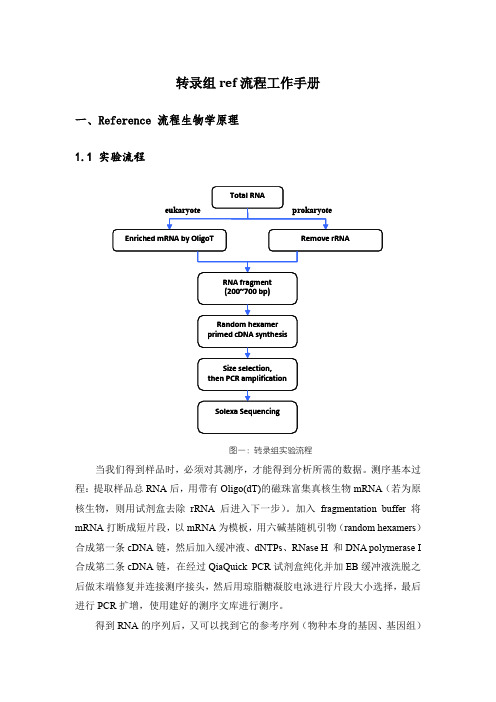
转录组ref流程工作手册一、Reference 流程生物学原理1.1 实验流程RNA fragment Random hexamer Size selection,prokaryoteeukaryoteTotal RNAEnriched mRNA by OligoT Remove rRNA(200~700 bp)primed cDNA synthesisthen PCR amplificationSolexa Sequencing图一:转录组实验流程当我们得到样品时,必须对其测序,才能得到分析所需的数据。
测序基本过程:提取样品总RNA后,用带有Oligo(dT)的磁珠富集真核生物mRNA(若为原核生物,则用试剂盒去除rRNA后进入下一步)。
加入fragmentation buffer将mRNA打断成短片段,以mRNA为模板,用六碱基随机引物(random hexamers)合成第一条cDNA链,然后加入缓冲液、dNTPs、RNase H 和DNA polymerase I 合成第二条cDNA链,在经过QiaQuick PCR试剂盒纯化并加EB缓冲液洗脱之后做末端修复并连接测序接头,然后用琼脂糖凝胶电泳进行片段大小选择,最后进行PCR扩增,使用建好的测序文库进行测序。
得到RNA的序列后,又可以找到它的参考序列(物种本身的基因、基因组)时,可以用reference流程对数据进行详细的分析。
Reference后面所有的流程都是基于参考序列进行的,所以选择正确的参考序列十分重要。
1.2信息分析流程得到测序序列后,即可利用比对软件,将所测序列比对到参考基因或基因组上,并进行后续分析,信息分析流程图如下:图二:转录组信息流程1.2.1原始fq序列简介测序得到的原始图像数据经base calling转化为序列数据,我们称之为raw data或raw reads,结果以fastq文件格式存储,fastq文件为用户得到的最原始文件,里面存储reads的序列以及reads的测序质量。
转录组测序数据分析(有参考基因组)

转录组测序数据分析(有参考基因组)一、数据分析流程二、数据分析内容1. 数据预处理目的:对原始测序数据进行一定程度的过滤。
原理:根据测序接头以及测序质量对原始的测序数据进行预处理,其中,测序质量Q与测序错误E之间的关系如下:结果:对预处理后质量以及碱基分布统计进行统计2. 比对基因组目的:将经过预处理的测序数据与参考基因组进行相似性比对。
原理:Burrower-Wheeler转换算法与splicing比对算法。
1)Burrower-Wheeler转换算法:由于测序数据量非常大,与整条基因组比对所需资源与时间是较为巨大的。
目前,我们采用Burrower-Wheeler(BWT)算法对基因进行建立索引、碱基压缩等过程,这样可以很大程度上加快比对速度,减少比对过程中所需资源。
2)splicing比对算法:即分段比对算法,当某条测序序列位于转录本剪切位点时,也就是这条序列同时属于两个外显子,如果将它与参考基因组进行比对,由于基因组两个外显子之间含有intron区,那么它将无法找到它合适的位置;但是应用分段比对算法就可以将这条测序序列分割变成多段子序列,然后应用这些段子序列与基因组进行比对,这样就可以找到它们真正的位置。
Vps28基因的一个分段比对的结果,蓝线连接的两端即为被分割的子序列,可见此种算法非常的适用于转录组测序。
结果展示:应用比对结果进行一些相关mapping统计,测序饱和度及测序5’,3’ bias统计。
Multi mapping,Unique mapping及Unique gene-body mapping统计。
饱和度分析,当reads达到一定测序量后,基因覆盖率基本达到饱和。
测序3’,5’偏好性统计,测序主要集中于基因bady区,两端偏向性较轻。
3. 基因表达水平研究目的:应用基因组比对结果进行基因定量。
原理:从指定物种基因模型(基因结构)中得到gene、exon、intron以及UTR等位置信息,通过基因组比对结果计算出在不用区域富集片段数目,然后应用RPKM/FPKM标准化公式对富集片段的数量进行归一化。
生物信息学的转录组数据分析

生物信息学的转录组数据分析一、引言转录组是一个生物组织或细胞中所有转录的RNA分子的总和,它反映了基因在特定条件下的表达水平。
转录组数据分析是生物信息学中的一个重要领域,它通过对转录组数据的处理和解读,可以揭示基因的功能和调控机制,以及在疾病发生发展中的作用。
本文将介绍转录组数据分析的基本步骤和方法。
二、数据预处理转录组数据通常以测序的形式存在,因此首先需要进行数据质控和预处理。
数据质控主要包括去除接头序列、低质量序列过滤、去除待测序列污染等步骤,以保证后续分析的准确性和可靠性。
预处理包括剔除低质量碱基、去除接头序列、剪切序列、质量修剪、构建序列库等步骤,以准备分析所需的干净数据。
三、基因表达分析基因表达分析是转录组数据分析的核心内容之一。
它通过比较不同条件下的基因表达水平,揭示基因的差异表达情况。
基因表达分析方法包括差异基因表达分析、基因聚类分析和基因富集分析等。
差异基因表达分析可以筛选出在不同条件下表达显著差异的基因,通过Gene Ontology(GO)和通路富集分析可以进一步了解这些差异基因的功能和相关通路。
四、基因调控网络分析基因调控网络分析是转录组数据分析的另一个重要方面。
它通过挖掘转录因子和靶基因之间的关系,揭示基因调控网络的结构和功能。
基因调控网络分析方法包括共表达网络分析和转录因子-靶基因分析等。
共表达网络分析可以用来发现与特定条件相关的基因模块,而转录因子-靶基因分析可以用来确定重要的转录因子并预测其功能。
五、功能注释与通路分析功能注释和通路分析是转录组数据分析的重要环节。
功能注释用于对差异表达基因进行功能注释,以了解其可能的生物学功能和参与的调控通路。
通路分析则是将差异基因映射到特定通路中,以揭示基因在特定生物学过程中的功能和相互作用关系。
功能注释和通路分析可以辅助我们理解基因调控网络的功能和调控机制。
六、数据可视化数据可视化是转录组数据分析的一个重要环节,它通过图表、散点图、热图等形式展示转录组数据的信息,增强数据分析结果的直观性和可解释性。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
v1.0 可编辑可修改一、生物信息分析流程获得原始测序序列(Sequenced Reads)后,在有相关物种参考序列或参考基因组的情况下,通过如下流程进行生物信息分析:二、项目结果说明1 原始序列数据高通量测序(如illumina HiSeq TM2000/MiSeq等测序平台)测序得到的原始图像数据文件经碱基识别(Base Calling)分析转化为原始测序序列(Sequenced Reads),我们称之为Raw Data或Raw Reads,结果以FASTQ(简称为fq)文件格式存储,其中包含测序序列(reads)的序列信息以及其对应的测序质量信息。
FASTQ格式文件中每个read由四行描述,如下:@EAS139:136:FC706VJ:2:2104:15343:197393 1:Y:18:ATCACGGCTCTTTGCCCTTCTCGTCGAAAATTGTCTCCTCATTCGAAACTTCTCTGT+@@CFFFDEHHHHFIJJJ@FHGIIIEHIIJBHHHIJJEGIIJJIGHIGHCCF其中第一行以“@”开头,随后为illumina 测序标识符(Sequence Identifiers)和描述文字(选择性部分);第二行是碱基序列;第三行以“+”开头,随后为illumina 测序标识符(选择性部分);第四行是对应序列的测序质量(Cock et al.)。
illumina 测序标识符详细信息如下:第四行中每个字符对应的ASCII值减去33,即为对应第二行碱基的测序质量值。
如果测序错误率用e表示,illumina HiSeq TM2000/MiSeq的碱基质量值用Qphred表示,则有下列关系:公式一:Qphred = -10log10(e)illumina Casava 版本测序错误率与测序质量值简明对应关系如下:2 测序数据质量评估测序错误率分布检查每个碱基测序错误率是通过测序Phred数值(Phred score, Q)通过公式1phred转化得到,而Phred 数值是在碱基识别(Base Calling)过程中通过一种预测碱基判别发生错误概率模型计算得到的,对应关系如下表所显示:illumina Casava 版本碱基识别与Phred分值之间的简明对应关系测序错误率与碱基质量有关,受测序仪本身、测序试剂、样品等多个因素共同影响。
对于RNA-seq技术,测序错误率分布具有两个特点:(1)测序错误率会随着测序序列(Sequenced Reads)的长度的增加而升高,这是由于测序过程中化学试剂的消耗而导致的,并且为illumina高通量测序平台都具有的特征(Erlich and Mitra, 2008; Jiang et al.)。
(2)前6个碱基的位置也会发生较高的测序错误率,而这个长度也正好等于在RNA-seq建库过程中反转录所需要的随机引物的长度。
所以推测前6个碱基测序错误率较高的原因为随机引物和RNA模版的不完全结合(Jiang et al.)。
测序错误率分布检查用于检测在测序长度范围内,有无异常的碱基位置存在高错误率,比如中间位置的碱基测序错误率显著高于其他位置。
一般情况下,每个碱基位置的测序错误率都应该低于%。
v1.0 可编辑可修改图测序错误率分布图横坐标为reads的碱基位置,纵坐标为单碱基错误率GC含量分布检查GC含量分布检查用于检测有无AT、GC 分离现象,而这种现象可能是测序或者建库所带来的,并且会影响后续的定量分析。
在illumina测序平台的转录组测序中,反转录成cDNA时所用的6bp 的随机引物会引起前几个位置的核苷酸组成存在一定的偏好性。
而这种偏好性与测序的物种和实验室环境无关,但会影响转录组测序的均一化程度(Hansen et al.)。
除此之外,理论上G和C碱基及A和T碱基含量每个测序循环上应分别相等,且整个测序过程稳定不变,呈水平线。
对于DGE测序来说,由于随机引物扩增偏差等原因,常常会导致在测序得到的每个read前6-7个碱基有较大的波动,这种波动属于正常情况。
v1.0 可编辑可修改图GC含量分布图横坐标为reads的碱基位置,纵坐标为单碱基所占的比例;不同颜色代表不同的碱基类型测序数据过滤测序得到的原始测序序列,里面含有带接头的、低质量的reads,为了保证信息分析质量,必须对raw reads进行过滤,得到clean reads,后续分析都基于clean reads。
数据处理的步骤如下:(1) 去除带接头(adapter)的reads;(2) 去除N(N表示无法确定碱基信息)的比例大于10%的reads;(3) 去除低质量reads。
RNA-seq 的接头(Adapter, Oligonucleotide sequences for TruSeq TM RNA and DNA Sample Prep Kits) 信息:RNA 5’ Adapter (RA5), part # :5’-AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT-3’RNA 3’ Adapter (RA3), part # :5’-GATCGGAAGAGCACACGTCTGAACTCCAGTCAC(6位index)ATCTCGTATGCCGTCTTCTGCTTG-3’v1.0 可编辑可修改图原始数据过滤结果测序数据质量情况汇总表数据产出质量情况一览表Sample name Raw reads Clean reads clean bases Error rate(%)Q20(%)Q30(%)GC content(%)数据质量情况详细内容如下:(1) Raw reads:统计原始序列数据,以四行为一个单位,统计每个文件的测序序列的个数。
(2) Clean reads:计算方法同 Raw Reads,只是统计的文件为过滤后的测序数据。
后续的生物信息分析都是基于Clean reads。
(3) Clean bases:测序序列的个数乘以测序序列的长度,并转化为以G为单位。
(4) Error rate:通过公式1计算得到。
(5) Q20、Q30:分别计算 Phred 数值大于20、30的碱基占总体碱基的百分比。
(6) GC content:计算碱基G和C的数量总和占总的碱基数量的百分比。
3 参考序列比对分析测序序列定位算法:根据不同的基因组的特征,我们选取相对合适的软件(动植物用TopHat(Trapnell et al., 2009)、真菌或者基因密度较高的物种用Bowtie),合适的参数设置(如最大的内含子长度,会根据已知的该物种的基因模型来进行统计分析),将过滤后的测序序列进行基因组定位分析。
下图为TopHatv1.0 可编辑可修改的算法示意图:Tophat的算法主要分为两个部分:(1) 将测序序列整段比对到外显子上。
(2) 将测序序列分段比对到两个外显子上。
我们统计了实验所产生的测序序列的定位个数(Total Mapped Reads)及其占clean reads的百分比,其中包括多个定位的测序序列个数(Multiple Mapped Reads)及其占总体(clean reads)的百分比,以及单个定位的测序序列个数(Uniquely Mapped Reads)及其占总体(clean reads)的百分比。
Reads与参考基因组比对情况统计表Reads与参考基因组比对情况一览表比对结果统计详细内容如下:(1) Total reads:测序序列经过测序数据过滤后的数量统计(Clean data)。
(2) Total mapped:能定位到基因组上的测序序列的数量的统计;一般情况下,如果不存在污染并且参考基因组选择合适的情况下,这部分数据的百分比大于 70%。
(3) Multiple mapped:在参考序列上有多个比对位置的测序序列的数量统计;这部分数据的百分比一般会小于10%。
(4) Uniquely mapped:在参考序列上有唯一比对位置的测序序列的数量统计。
(5) Reads map to '+',Reads map to '-':测序序列比对到基因组上正链和负链的统计。
(6) Splice reads:(2)中,分段比对到两个外显子上的测序序列(也称为Junction reads)的统计,Non-splice reads为整段比对到外显子的将测序序列的统计,Splice reads的百分比取决于测序片段的长度。
Reads在参考基因组不同区域的分布情况对Total mapped reads的比对到基因组上的各个部分的情况进行统计,定位区域分为Exon(外显子)、Intron(内含子)和Intergenic(基因间隔区域)。
正常情况下,Exon (外显子)区域的测序序列定位的百分比含量应该最高,定位到Intron (内含子) 区域的测序序列可能是由于非成熟的mRNA的污染或者基因组注释不完全导致的,而定位到Intergenic(基因间隔区域)的测序序列可能是因为基因组注释不完全以及背景噪音。
图Reads在参考基因组不同区域的分布情况Reads在染色体上的密度分布情况对Total mapped reads的比对到基因组上的各个染色体(分正负链)的密度进行统计,如下图所示,具体作图的方法为用滑动窗口(window size)为1K,计算窗口内部比对到碱基位置上的reads的中位数,并转化成 log。
正常情况2下,整个染色体长度越长,该染色体内部定位的reads总数会越多(Marquez et al.)。
从定位到染色体上的reads数与染色体长度的关系图中,可以更加直观看出染色体长度和reads总数的关系。
图Reads在染色体上的密度分布图上图:横坐标为染色体的长度信息(以百万碱基为单位),纵坐标为log2(reads的密度的中位数),绿色为正链,红色为负链下图:横坐标为染色体的长度信息(单位为Mb),纵坐标为mapped到染色体上的reads数(单位为M)Reads比对结果可视化我们提供RNA-seq Reads在基因组上比对结果的bam格式文件,部分物种还提供相应的参考基因组和注释文件,并推荐使用IGV (Integrative Genomics Viewer) 浏览器对bam文件进行可视化浏览。
IGV浏览器具有以下特点:(1)能在不同尺度下显示单个或多个读段在基因组上的位置,包括读段在各个染色体上的分布情况和在注释的外显子、内含子、剪接接合区、基因间区的分布情况等;(2)能在不同尺度下显示不同区域的读段丰度,以反映不同区域的转录水平;(3)能显示基因及其剪接异构体的注释信息;(4)能显示其他注释信息;(5)既可以从远程服务器端下载各种注释信息,又可以从本地加载注释信息。