精细有机合成 总结
精细有机合成—构成环状化合物的反应

由Knorr合成法得到的产物可水解脱羧,生成取代吡咯。许多其 他吡咯,尤其是用于卟啉合成中的吡咯,也是利用这一方法制备, 不过改变不同的取代基而已。为了方便地脱去烷氧羰基,在Knorr合 成中,用苯基和叔丁基酯取代了乙酯基。如原料中的氨苯上有烃基, 则可得到N-烃基吡咯。
α,β-不饱和羰基化合物是极活泼的亲二烯体系,并且代表了该合成方 法中最有价值的组分,其典型的例子有丙烯醛、丙烯酸及其酯、顺丁烯 二酸及其酸酐和丁炔二酸:
+ +
+
+
(2) Robinson增环反应 活泼亚甲基化合物与α,β-不饱和酮、酯、腈等起Michael反应,然后起
醇醛缩合反应称之为Robinson增环反应,常用于合成环状化合物。在合 成六元环烃,特别在甾体化合物的合成上具有重要作用。这种方法分两个 阶段进行。先起Michael加成反应,接着起分子内的羟醛缩合反应,增环 生成环己酮。一般采用催化量的碱,主要得到1,4-加成产物,采用当量碱 则主要得到环合产物.这样可以利用两步合一的反应方便地合成六元环。
二卤环丙烷用AgNO3处理,可转化为烯丙基化合物,这是用卡 宾增长碳链的另一种方法。
卡宾与杂环体系的烯键加成,形成扩环产物,这在合成上十分有用:
3.1.2 四元环衍生物 用1,3-二卤代烷对活性亚甲基化合物进行分子内烷基化,例如
在强碱存在下,丙二酸酯与1,3二溴丙烷成环,生成环丁烷衍生物。
四元环除由丙二酸酯法合成外,还可以由[2+2]环加成反应合成。[2+2] 环加成是由两个烯分子组成四元环的反应。简单的烯烃在加热时不能生成 环丁烷衍生物,丙烯腈容易二聚成顺-和反-1,2—二氰基环丁烷:
精细有机合成单元反应复习.docx

精细有机合成单元反应复习第一章精细有机合成基础知识要点一、取代基效应1、诱导效应1)因为成键原子的电负性差异而引起的电荷传递作用2)所有取代基都存在诱导效应,即诱导效应无处不在3)了解诱导效应强弱、方向是认识该效应的核心2、共辄效应1)了解共觇效应的类型、能够判断分子或屮间体是否存在共觇效应2)共辘效应影响的结果是因为电子离域或共享而使分子、活性中间体稳定。
如正离子、负离子或自由基都能够被共辘所稳定。
3)共轨效应方向受诱导影响如分析下面分子或活性中间体的共轨方向H2C^=C—C^=CH2H2C^=C—H3C—C CH—c—OC2H5H H H3、空间效应1)空间效应产生原因一般有两种:A因为取代基体积而产生的空间位阻;B因为分子的刚性结构或键角张力2)与诱导效应一样,所有分子都可能存在空间效应3)空间效应对分子或活性中间体的影响可能是起促进或阻碍作用二、有机反应历程1、脂肪族取代反应重点了解亲核取代反应的影响因素2、芳香族亲电取代了解一般反应历程、取代反应的定位是重点(包括多取代基存在下的取代反应、稠环的单取多取代)3、芳香族亲核取代了解影响亲核収代的影响因素特别是环上已有収代基存在下对亲核取代的活性影响三、溶剂1、了解溶剂的分类2、溶剂对反应活性的影响Huges-Ingold规则3、了解非质子传递性溶剂对亲核反应的影响例题:作为亲核试剂,卤素阴离子在一般极性溶剂屮亲核活性顺序如何?在非质子传递性溶剂中亲核性顺序又如何?试对此作出合理解释。
考占.P 八、、•1、収代基效应的判断:下面反应物或活性中间体存在共轨效应的是()2、极性溶剂对下面反应有活化作用的是()3、下面芳香族亲核取代反应活性最高的是()4、下面亲核试剂亲核性最强的是()5、下面自由基稳定性最好的是()6、在水中卤素阴离子亲核性顺序是:在DMF溶剂中亲核性顺序是:解释其原因?7本章习题28、芳香族亲电取代定位效应第二章卤代反应知识要点一、芳香族环上卤代反应1、亲电反应历程、酸催化反应或无需催化2、连串反应(即有多卤代反应),控制反应产物的关键是卤化反应深度3、反应有较弱的可逆性4、反应的一般条件:反应溶剂:反应物本体、有机溶剂、酸、水反应试剂:单质、次卤酸、NCS、NBS、HX+氧化剂反应活性:与反应物有关、与试剂有关、与溶剂有关主要反应:苯卤代、甲苯卤代、苯酚卤代、苯胺卤代5、碘代反应的特殊性二、不饱和桂的加成卤代1、加成反应活性都很高,在温和的条件下都能够实现加成2、加成方向马氏规则(本质上是取代基效应)3、加成试剂HX、X2、HOX4、共觇烯坯的加成1, 2■加成比1, 4■加成活性更高,所以反应可能首先生成1, 2■加成产物,但1, 4■加成产物稳定性更好,较高的反应温度或较反的反应时间,1, 4■加成产物可能会成为主要产物三、烯丙位或侧链的自由基卤代1、反应是自由基历程,反应条件光照或加热,引发剂能够促进反应2、当有多个位置可能被取代时,节位或烯丙位优先3、反应有多取代异构体,由卤化深度的控制4、侧链卤代吋,不能有过渡金属离子存在(即Lewis酸),否则可能有环上卤代发生5、烯丙位卤代反应的竞争是双键的加成,高温不会发生加成,但低温下反应,主要产物是加成产物四、疑基置换反应1、醇的置换卤代反应活性:短链〉长链、叔醇〉仲醇〉伯醇醇〉酚卤代试剂:浓HX酸-浓盐酸、浓蛍澳酸(HI不宜)含磷卤代试剂・PX3、PX5含硫卤代试剂-SOCb2、酚的置换卤代只能用强卤代试剂如PX3、PX.53、酸的卤代制备酰卤卤代试剂一般用SOCB、PX3五、拨基a—位卤代1、反应是指醛、酮、酸衍生物的。
有机合成工个人工作总结

有机合成工个人工作总结在过去的一年中,我作为有机合成工在公司的工作经历让我收获颇丰。
首先,我在有机化合物合成方面已经有了很多经验,从简单的反应到复杂的合成路线,我都能够独立完成。
这些经验让我对有机化合物的合成有了更深的理解,并且提高了我的化学实验技能。
其次,我在团队合作方面也取得了进步。
在项目合作中,我和同事们密切合作,及时沟通,互相协助,最大限度地提高了工作效率。
在团队合作中,我学会了克服自己的个人局限,思考问题更全面,更能够站在整个团队的利益角度思考问题。
除此之外,我还在实验仪器操作方面有了一定的提高。
在过去的一年中,我参与了多种实验仪器的操作和维护工作,不断地提高自己的实验技能和仪器使用技巧,确保了实验数据的准确性和可靠性。
总结来说,过去一年的工作经历让我在有机合成领域和团队合作方面取得了很多进步。
在未来的工作中,我将继续努力提高自己的专业技能和团队合作能力,为公司的发展贡献更大的力量。
在过去的一年中,我作为有机合成工在公司的工作经历让我收获颇丰。
首先,我在有机化合物合成方面已经有了很多经验,从简单的反应到复杂的合成路线,我都能够独立完成。
这些经验让我对有机化合物的合成有了更深的理解,并且提高了我的化学实验技能。
在过去的一年里,我的工作主要集中在设计和优化有机合成路线。
我积极参与了多个项目,并且成功完成了一些复杂有机分子的合成。
在实验过程中,我不断探索新的合成方法和反应条件,不断地优化合成路线,提高了合成产物的产率和纯度。
在这个过程中,我学到了很多从实验设计到实际操作的经验,也积累了很多有机合成的技能和知识。
另外,我也参与了实验数据分析和结构表征的工作。
我使用了各种仪器对合成产物进行了表征和分析,包括NMR、IR、MS 等技术。
通过仪器分析,我验证了合成产物的结构和纯度,并及时发现了反应中的一些问题。
这些实验数据为后续的研究和开发工作提供了重要的支持。
除了个人技能的提高之外,我也意识到团队合作的重要性。
有机合成实验工作总结报告

有机合成实验工作总结报告有机合成实验是化学专业学生必须进行的一项重要实践操作。
通过实验操作,我对有机合成反应有了更深刻的理解,并掌握了有机合成实验的基本方法和技能。
在本次实验中,我参与了安全操作、实验记录、化合物合成等多个方面的工作。
首先,在实验过程中,我非常重视实验室的安全问题。
在进入实验室之前,我仔细阅读了实验的操作指南,并根据实验室安全规定,戴上了实验室必要的防护装备。
实验过程中,我十分注重实验室的卫生,及时清理实验台面上的废弃物,并注意实验废液的正确处理。
同时,在操作过程中,我严格遵循实验操作规程,确保实验中的化学品的正确使用和储存,保证了实验的顺利进行。
其次,我积极参与了实验数据的记录工作。
在实验中,我认真地观察和记录实验过程中的现象和变化,并使用适当的图表和表格来展示实验数据。
在记录实验数据时,我十分注重数据的准确性和完整性。
同时,在实验过程中,我注意及时与同组同学、助教和教员进行实验数据的交流和讨论,共同解决实验中遇到的问题。
最后,我参与了有机合成化合物的合成工作。
在实验中,我根据实验指南中的合成路线图,精确计算了化合物的摩尔比例和反应物的用量。
在合成工作中,我注重化学反应条件的控制,保证反应的顺利进行。
同时,在合成过程中,我注意观察反应物是否完全转化,及时进行反应时间的调整。
当反应完成后,我采用适当的技术手段对产物进行分离和提取,并使用相关仪器对合成的化合物进行表征。
通过本次有机合成实验,我不仅提高了实验技能和安全意识,还加深了对有机化学反应机理的理解。
同时,通过实验记录和数据处理,我学会了如何准确地记录实验数据和分析实验结果。
我相信这次实验经历对我今后的科研工作和学习都具有积极的影响。
精细有机合成技术:酯的缩合

• 在无水条件下,使用活性更强的碱(如RONa、NaNH2 等)作催化剂,两分子的酯就会通过消除一分子的醇缩 合在一起。
反应历程:在催化剂乙醇钠的作用下,酯先生成负碳离 子,并向另一分子酯的羰基碳原子进行亲核进攻,得初 始加成物;初始加成物消除烷氧负离子,生成β-酮酸酯。
➢ 含有活泼α-H的酯均可发生自身缩合反应。 ➢ 当含两个或三个活泼α-H的酯缩合时,产物β-酮酸酯的
感谢观看
酯缩合反应在非质子溶剂中进行比较顺利。常用的 溶剂有乙醚、四氢呋喃、乙二醇二甲醚、苯及其同系物, 二甲基亚砜(DMSO)、二甲基甲酰胺(DMF)等。有些反 应也可以不用溶剂。酯合反应需在无水条件下完成,这 是由于催化剂遇水容易分解并有氢氧化钠(游离碱)生 成,后者可使酯水解皂化,从而影响酯缩合反应进行。
精细有机合成技术
目
录
酯的缩合
酯-酯缩合 1
Contents
酯-酮缩合 2
酯-酮缩合
酯与酮在碱性条件下缩合,生成具有两个羰基的β二酮类化合物。其反应与酯酯缩合反应相似。由于酮的 α-H活性比酯大,在碱性条件下,酮比酯更易脱去质子, 酮形成的负碳离子向酯羰基进行亲核加成而生成产物。
• 如丙酮、草酸二乙酯和甲醇钠的甲醇溶液按1:1:1的 摩尔比反应,经酸化得2,4-二酮戊酸乙酯。
通常,酮的结构越复杂,反应活性往往越弱。含活 泼α-H的不对称酮与酯缩合时,取代基较少的α-碳形成 负离子,向酯进行亲核加成。若酮分子中仅一个α-碳上 有氢原子,或酯不含活泼α-H,产物都比较单纯。如:
• 如果酯的反应活性太低,则可能发生酮酮自身缩合副反 应。若酯的α-H的酸性较酮α-H高,则可能发生酯酯自 身缩合和诺文葛耳一多布纳副反应。
酸性比醇大得多,在有足够量的醇钠等碱性催化剂作 用下,产物几乎可以全部转化成稳定的β-酮酸酯钠盐, 从而使反应平衡向右移动。
精细有机化学合成
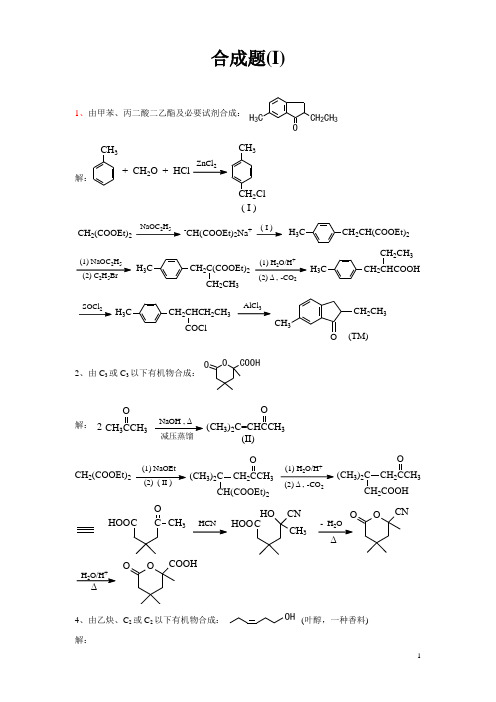
合成题(I)1、由甲苯、丙二酸二乙酯及必要试剂合成:OH 3CCH 2CH 3解:CH 3CH 3CH 2Cl + CH 2O + HCl2( I )2+2CH 2(COOEt)225-CH(COOEt)2Na +( I )H 3C CH 2CH(COOEt)22525H 3C CH 2C(COOEt)22CH 3H 3CCH 2CH 2CH 32H 3CCH 2CHCH 2CH 33CH 3OCH 2CH 3(TM)2、由C 3或C 3以下有机物合成:O COOHO解: CH 33ONaOH , ∆2(CH 3)23O(II)CH 2(COOEt)2(2) ( II )(1) NaOEt (CH 3)2C CH 23OCH(COOEt)22+2(CH 3)2CCH 23OCH 2COOHOO CN2CC CH 3OHOO CHO CN CH 3HOOH 2O/H ∆OO COOH4、由乙炔、C 2或C 2以下有机物合成:OH (叶醇,一种香料)解:CH 3CH 2CCMgBrO 干醚H 2O CH 3CH 2C CCH 2CH 2OHH 2C=CHHCH 2CH 2OH CH 3CH 2CH CHCH CNaCH CCH 2CH 33C H Br25(TM)5、由Br 为有机原料合成:COOCH 3 解:BrNaCNCNH 2O COOHCOClCHO2H /Pd-BaSOBr COOHCOOCH 3Br3+4NaOH/醇COOCH 3ZnBrCHO干醚H 2O COOCH 3- H 2O COOCH3(TM)6、由O 合成3解:OCH 3OHCH 3CH MgI H 2O 24(1) B H 22CH 3HH (TM)7、由丙二酸二乙酯合成COO -N +H 3解:Br Br CH(COOEt)2CH 2(COOEt)2NH O KOHNK O NCH(COOEt)2O(1) C H ONa 322NC(COOEt)2OOCH 2CH(CH 3)22+2(CH 3)2CHCH 2CHCOOH2NH 3COO -+即:8、由苯合成ICl Cl Cl解:NO 2NH 2NHCOCH 3HNO 3H 2SO 432Fe + HClNHCOCH 3NH 2NHCOCH 32Cl ClFe + HCl2NaNO 2+过量HCl。
有机合成实验工作总结
有机合成实验工作总结引言有机合成实验是化学领域中重要的研究内容之一,通过有机合成实验,可以合成出各种有机化合物,并进一步研究它们的物理和化学性质。
本文将对我在实验室进行的有机合成实验工作进行总结,并对实验过程中遇到的问题及解决方法进行探讨。
实验目的本次有机合成实验的目的是合成一种特定的有机化合物,并通过对合成产物的表征,验证合成反应的成功与否。
同时,通过实验过程,提高我对有机化合物合成的实际操作能力和实验技术水平。
实验过程1. 实验材料准备在实验开始前,首先需要准备实验所需的各种材料和试剂。
本次实验要求的材料有:•反应容器:玻璃烧杯、试管、漏斗等;•实验试剂:氯化亚砜、甲醛、氢氧化钠等;•仪器设备:加热装置、搅拌器等。
2. 实验步骤本次实验的合成反应是一个多步反应,需要经过数个化学步骤才能最终得到目标产物。
以下是本次实验的主要步骤:•步骤1:制备反应物溶液。
将氯化亚砜溶解在有机溶剂中,并加入适量的甲醛。
•步骤2:加热反应。
将反应溶液经过加热,并同时不断搅拌,以保证反应的均匀进行。
•步骤3:中和反应。
在反应溶液中加入适量的氢氧化钠,使其中和反应达到最佳状态。
•步骤4:提取产物。
将反应结束后的溶液进行提取,得到目标产物。
3. 反应结果与分析通过对实验得到的产物进行表征和分析,可以判断实验的成功与否,以及产物的纯度和结构。
本次实验产物的分析结果如下:•红外光谱:产物在红外光谱上显示出了特定的吸收峰,与合成目标相符合。
•核磁共振谱:通过核磁共振谱分析,确认了产物的分子结构和官能团。
实验中的问题与解决在实验过程中,我遇到了一些问题,通过及时的解决和改进,最终完成了实验的目标。
以下是我在实验中遇到的问题及解决方法:1.产物纯度不高:通过反复提取、结晶等方法,提高产物的纯度。
2.反应效率低下:改变反应条件、优化实验步骤,以提高反应的效率。
3.实验设备不足:与同学合作使用设备、或者提前预约使用设备。
实验心得与收获通过参与本次有机合成实验,我收获了以下方面的经验和教训:1.实验技术的提高:通过实际操作,我提高了实验技术的能力,掌握了更多的实验技巧。
精细有机合成知识点总结
精细有机合成知识点总结一、有机合成基础知识1. 有机化合物的结构特点:有机化合物以碳为主要元素,通常含有氢、氧、氮、硫等元素,具有复杂的结构和多样的性质。
有机化合物的结构特点对于合成时的反应条件和合成路径具有重要的影响。
2. 有机合成的基本原理:有机合成是指通过将简单的有机化合物经过一系列的反应转化成目标化合物的过程。
合成的基本原理包括合成途径的选择、反应条件的控制、反应机理的理解等方面。
3. 有机合成的分类:有机合成可以根据合成途径、合成目标、合成方法等多个方面进行分类。
常见的分类包括:官能团化合成、碳碳键形成、环化反应、取代反应等。
二、精细有机合成的理论基础1. 反应机理:在精细有机合成中,对于反应的机理的理解是非常重要的。
包括反应物的选择、反应条件的控制、中间体的形成等方面的理论基础。
2. 功能团保护和去保护:在有机合成过程中,有时需要对特定的官能团进行保护,以防止其在反应过程中发生不必要的改变。
同时,也需要在合成的适当时机去除这些保护基团,以获得目标产物。
3. 立体化学:有机合成中的立体化学是一个重要的理论基础。
包括立体化学的理论基础、手性分子的制备和合成、手性识别和手性分离等方面的知识。
4. 共价键断裂和形成:在有机合成中,共价键的断裂和形成是非常常见的反应过程。
了解这些反应的机理和条件对于合成路径的选择和优化具有重要的意义。
三、精细有机合成的实验技术1. 反应条件的控制:在实际合成过程中,对反应条件的控制是非常重要的。
包括温度、压力、溶剂的选择等方面的实验技术。
2. 操作技术:精细有机合成涉及到很多精细的操作技术,包括溶剂的蒸馏、试剂的使用、产物的提取和纯化等。
3. 合成路径的选择和优化:在精细有机合成中,选择合适的合成路径对于提高产物收率和纯度都具有重要的意义。
需要根据反应物的结构特点和反应机理进行合适的路径设计和优化。
四、精细有机合成的应用1. 药物合成:精细有机合成在药物合成领域有着广泛的应用。
精细有机合成化学及工艺学
精细有机合成化学及工艺学1. 精细有机合成化学有机合成化学是化学的一个分支领域,主要是研究有机物的合成方法、合成反应机理、合成原料等。
而精细有机合成化学则是有机合成化学的一个重要分支,它研究的是更加复杂的有机分子的合成方法以及高效的制备技术。
1.1 精细有机合成化学的概念精细有机合成化学指的是合成更加复杂的有机化合物的方法,例如生物活性分子、药物分子、天然产物等。
它不仅需要充分了解化学反应的机理,还需要考虑反应的条件、催化剂的选择以及中间体的稳定性等因素,从而使得反应具有高效、高收率、高纯度等特点。
1.2 精细有机合成化学的原则在精细有机合成化学中,以下原则是非常重要的:•合成路线的选择。
在合成复杂有机分子的过程中,由于反应底物的复杂性和特殊性,合成路线的选择是非常重要的。
需要进行多次的尝试和改进,找到最优的合成路线。
•反应条件的优化。
精细有机合成化学涉及到多个反应步骤,需要对反应条件进行优化,例如温度、压力、溶剂的选择等,以保证反应的效率和产率。
•保护基团的选择。
在有机化合物合成中,由于末端基团的敏感性,需要采用保护基团以防止它们在反应过程中受到损伤或者分解。
•催化剂的选择和使用。
精细有机合成化学中催化剂的选择和使用非常重要,可以大大提高反应的效率和速率,以及降低反应的温度和压力等。
2. 精细有机合成工艺学精细有机合成工艺学是精细有机合成化学的实践应用,主要研究有机化合物的制备工艺,包括生产过程的设计、合成路线的开发、分离和纯化技术等,以实现工业化大规模生产。
2.1 精细有机合成工艺学的应用领域精细有机合成工艺学广泛应用于生产中,包括制药、农药、色素、涂料、环保材料等领域。
•制药领域。
精细有机合成工艺学在药物合成中亦有突出作用。
现代药物的设计和化学合成往往需要使用多步、多中间体和多催化剂等工艺,因此精细有机合成工艺学在药物生产中显得格外重要。
•农药领域。
在农药的研发和生产中,精细有机合成工艺学也扮演了重要角色。
有机合成实验工作总结范文
有机合成实验工作总结范文
有机合成实验工作总结。
在有机合成实验工作中,我们通过一系列化学反应,将简单的有机化合物转化
为复杂的有机分子。
这项工作需要精确的操作技巧和深厚的化学知识。
在最近的一次实验中,我们成功地合成了目标化合物,并取得了令人满意的结果。
首先,我们进行了反应物的选择和准备。
通过仔细筛选和精确称量,我们确保
了反应物的纯度和量的准确性。
接下来,我们进行了反应条件的优化。
通过调整温度、溶剂和催化剂的选择,我们最终找到了最适合的反应条件,使得目标化合物的产率和纯度都得到了提高。
在反应进行过程中,我们密切观察了反应的进行,并及时调整了反应条件,以
确保反应的顺利进行。
最终,我们成功地得到了目标化合物,并通过NMR、IR等
技术手段对其进行了表征。
结果表明,我们所合成的化合物符合预期结构,且纯度较高。
在实验过程中,我们也遇到了一些困难和挑战。
例如,某些反应物的选择和合
成步骤的优化都需要耗费大量的时间和精力。
但是,通过团队的合作和不懈的努力,我们最终克服了这些困难,取得了成功。
总的来说,这次实验工作为我们提供了宝贵的经验和教训。
我们不仅学会了如
何进行有机合成实验,还提高了实验操作的技能和化学知识。
通过这次实验,我们对有机合成的原理和方法有了更深入的理解,也为今后的研究工作奠定了坚实的基础。
我们相信,通过不断的努力和实践,我们将能够在有机合成领域取得更多的成果。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
第1章 绪论/1、精细化工及相关行业的概念初始原料:煤、石油、天然气、生物有机质(农林副产) 基础有机原料:乙烯、丙烯、丁二烯、苯、(甲苯)、二甲苯、(乙炔、萘)、合成气(CO + H2)等。
2.2 亲电取代反应→ 2.2.3 芳香族亲电取代定位规律→(1)影响定位的主要因素 2.2.3 芳香族亲电取代定位规律 (1) 影响定位的主要因素■ 已有取代基的性质: ①极性效应 ②空间效应■ 亲电试剂的性质——也包括: ①极性效应 ②空间效应 ■ 反应条件:主要-温度、催化剂和溶剂。
上述因素中,最重要的是已有取代基的极性效应。
芳香取代反应中,苯系亲电取代反应研究的最多,也最重要。
2.2 亲电取代反应→ 2.2.3 芳香族亲电取代定位规律→(2)两类定位基 (2)两类定位基已有取代基 Z 对新取代基 E 的定位作用有两种:■第一类定位基 邻、对位定位基:-O-、-N(CH3)3、-NH2、-OH 、-OCH3、-NHCOCH3、-OCOCH3、-F 、-Cl 、-Br 、 -I 、 -CH3、-CH2Cl 、-CH2COOH 、-CH2F 等。
■第二类定位基间位定位基:-N+(CH3)3、-CF3、 -NO2、-C≡N 、 -SO3H 、-COOH 、-CHO 、-COOCH3、-COCH3、-CONH2、-N+H3、-CCl3。
上节回顾 2.2 亲电取代反应芳香族亲电取代反应历程大多数亲电取代反应是按照经过σ配合物中间产物的两步历程进行的。
其通式如下:2.3 亲核取代反应→ 2.3.2 反应影响因素→(4)溶剂的影响 (4)溶剂的影响SN1反应的第1步是一个中性的化合物离解为两个带有不同电荷的离子,因此极性溶剂有利于反应的进行。
SN2反应中,因为极性溶剂与亲核试剂可以形成氢键,亲核试剂与反应物形成过渡态时,必须首先消耗能量破坏氢键,所以反应在不形成氢键的溶剂中进行,反应速度较快。
2.4 消除反应→ 2.4.3 影响消除反应的因素→(2)反应条件的影响■ 温度的影响:提高温度有利于消除反应。
6.2.1 硝化剂的活泼质点硝化剂:硝化反应中能够生成硝基正离子(NO2+)的试剂。
NO2+是亲电硝化反应的真正进攻质点。
具有X -NO2的化合物,可产生NO2+离子: 离解的难易程度,决定于 X -NO2 分子中X 的吸电子能力:① X 吸电子能力越强,越易形成 NO2+ 离子,硝化能力也愈强。
② X 吸电子能力的大小可由 X- 的共轭酸的酸度来表示。
Z 邻位E Z对位E Z间位E X NO 2 X - + NO 2+A r + H Ek 2 ArE + H+ Ar -H + E + k 1 k -1 A r + H E σ配合物 第一步 第二步6.2.3 硝化反应历程 ⑴ 混酸硝化研究表明混酸硝化机理① NO2+ 向芳烃发生亲电攻击生成π络合物② π络合物转化为σ络合物(控制阶段)③ 脱去质子得到硝化产物⑵ 活泼芳烃用硝基盐硝化硝化剂:硝基硼氟酸盐(NO2+BF4-)特 点:硝化能力很强,可不考虑NO2+生成速度对反应的影响⑶ 稀硝酸硝化一些活泼芳烃(酚、酚醚、某些N-酰基芳胺)常用稀硝酸作硝化剂。
动力学研究表明, υ = k [ArH][HNO2] 亚硝化阶段:① 芳烃首先与 亚硝酸 作用生成亚硝基化合物② 硝酸氧化 亚硝基化合物,硝酸本身被还原,重又生成亚硝酸 ⑷ 其它硝化\① 在醋酐中硝化① 在醋酐中硝化在已有取代基位置上,对(除 H 外)基团的取代硝化反应。
③ 有汞存在时的氧化硝化苯在汞的存在下,以较稀的硝酸进行硝化,同时可得硝基苯酚、二硝基苯酚、苦味酸等。
① D.V .S. 值大,表明硫酸浓度大,硝化能力强,适用于难硝化的物质; ② D.V .S. 值小,表明硫酸浓度小,硝化能力弱,适用于易硝化的物质 ⑵ 废酸计算浓度(硝化活性因数)( F.N.A .) 混酸硝化结束时,废酸中硫酸的计算浓度。
若以100 份混酸为计算基准,当ф≈1时:当ф≈1 时, F.N.A . 与 D.V.S. 的互换关系:当S 、N 为变量,F.N.A.为常数时,为一直线方程,说明满足相同废酸浓度的混酸组成可以是多样的,但有实际意义的混酸组成,仅是其中的一小段。
NO 2++ NO 2+NO 2+慢H NO 2H NO 2快NO 2+ H +NO 2++ NO 2+慢π络合物∞H ∞H NO 2快HNO 2H NO 2+ H + ArH + HNO 2 ArNO + H 2OArNO + HNO 3 ArNO 2 + HNO 2 NO 2+ HAc + H + + CH 3C OH ONO 2R + Y +Y R Y++ R + 废酸中水的重量废酸中硫酸的重量=...S V D NS N S A N F -=⨯-=14014010075100...N N 726318=⨯=反应生成的水7510072100N N N -=+-=废酸重量...100......A N F A N F S V D -=100...1......⨯+=S V D S V D A N F (140)140A N F NS ⨯-=6.4.2 配酸工艺 配酸时应注意:① 有及时导出热量的冷却装置,一般要求控制温度低于40 ℃,以减少硝酸的挥发和分解; ② 有效的混合装置; ③ 有效的设备的防腐措施;配酸方法:①间歇配酸法:生产能力低,适用于小批量多品种的生产 ②连续配酸法:生产能力大,适用于大吨位的生产由于浓硫酸遇水放出大量热量,应缓慢加入水中或废稀酸中,最后在40 ℃条件下先慢后快加硝酸并注意安全。
4.1 芳环上的取代氯化芳环上的取代氯化:在催化剂存在下,芳烃上的的 H 被 Cl 取代的过程(连串反应)。
ArH + X2 → ArX + HX (亲电取代) 4.1.1 反应理论⑴ 催化剂存在下氯气的氯化FeCl3、AlCl3、ZnCl2、MnCl2、SnCl4、TiCl4等路易斯酸红棕色液体(4) 酸催化下的次氯酸的氯化(5)二氯硫酰4.1.2 反应动力学 苯环上的氯化是一连串反应:室温下 k1 / k2 = 10,k3 很小,可忽略,所以在氯化反应前期,连串反应中三氯苯生成极少。
4.1.3 影 响 因 素(1)氯化深度(2)操作方式(3)反应温度(4)原料纯度(5)催化剂的选择 (6)反应介质⑴ 氯化深度环上取代氯化为连串反应。
氯化深度用参加氯化反应的原料的百分数来表示。
苯的一氯化反应速度常数,在常温下仅比二氯化反应速度常数大8.5倍左右,因此制备一氯化物时必须严格控制氯化深度。
如果要生成二氯化物,可以提高反应深度。
用反应液相对密度来控制。
氯苯用途比二氯苯大,反应中为增加氯苯含量,而减少多氯苯含量,可降低氯化反应的浓度,即减少苯的反应。
但剩余苯越多,则回收的苯越多,操作费用及损耗将增加,设备生成能力亦下降,因此要慎重选择反应浓度。
可由出口处氯化液的相对密度来控制氯化深度(不同的氯化液组成有各自的相对密度)。
(2) H2SO4为催化剂 H 2SO 4 H + + HSO 4-H + + Cl 2 Cl ++ HCl I 2 + Cl 2 2ICl ICl I + + Cl -I + + Cl 2 ICl + Cl +(3) I2为催化剂 FeCl 3 + Cl 2 [Cl +FeCl 4-] + [Cl +FeCl 4-]慢 k 1H Cl + FeCl 4-快 k 2Cl + HCl + FeCl 3SO 2Cl 2 ClSO 2- + Cl +Cl - + SO 2 C 6H 6 + Cl 2C 6H 5Cl + HCl C 6H 5Cl + Cl 2C 6H 4Cl 2 + HClυ1κ2υ2κ1C 6H 4Cl 2 + Cl 2C 6H 3Cl 3 + HCl κ3υ3HOCl H 2O +Cl Cl +H +快- H 2O Ar H Cl ( ) +ArCl ArH - H +快⑵ 操作方式操作可分为间歇式和连续式(单级塔式、多级槽式)。
连续、间歇过程主要差异是产物可能返回到反应区。
⑶ 反应温度早期氯苯的生产中,温度尽可能控制反应在35~40℃下进行,是为了防止二氯苯的生成过多。
近代,普遍采用氯化液的沸腾温度78~80℃下进行,用塔式反应器或列管式氯化器进行。
其他卤化反应的温度,要根据反应物的活泼性而定。
(5) 催化剂的选择在芳环上有较强的供电基(如羟基、氨基),在卤化时一般不用催化剂。
对活泼性较低的芳烃,一般要加入金属卤化物作催化剂。
对不活泼的芳烃的直接卤化,则要求有强烈的卤化条件和催化剂,一般用浓硫酸、碘或氯化碘。
(6) 反应介质 ① 水适 用:被氯化物和产物都是固体,且氯化反应较易进行,把被氯化物以很细的颗粒悬浮于水介质中,酸存在下氯化。
例,对硝基苯胺的氯化 ② 无机溶剂适 用:难溶于水中的有机物,常用的溶剂是硫酸。
例,蒽醌在浓硫酸介质中可直接氯化制1,4,5,8-四氯蒽醌 ③ 有机溶剂适 用:某些有机物的氯化可在有机溶剂(惰性)中进行。
例,萘的氯化以氯苯为溶剂;水杨酸的氯化以乙酸为溶剂;苯胺的氯化以水-醋酸为溶剂,随体系中水的增加,苯胺氯化速度增加。
(溶剂极性对芳香化合物氯化速度有影响。
) 4.2.2 甲苯的侧链氯化甲苯的侧链氯化是在光或引发剂引发下,采用沸腾氯化法进行的。
反应在塔式氯化器中进行,生产流程与氯苯的生产相近,反应中应避免金属存在,以防止环上取代氯化发生。
氯化反应完毕后通入少量空气吹走 HCl ,接着进行分馏操作。
分馏前进行洗涤或碱液中和是不适宜的,因为甲苯侧链氯化衍生物非常容易水解。
分馏设备应是搪瓷或搪玻璃,因为设备中存在铁质会引起产品缩合生成树脂状物。
近年来,也有用活性炭作催化剂使甲苯在300 ℃进行气相侧链氯化,甲苯的转化率达70 %,主要产物为苯氯甲烷;也有使甲苯在甲酰胺和偶氮二异丁腈存在下于75 ℃ ~ 85 ℃进行反应,以提高甲苯侧链氯化的转化率和选择性。
乙苯、异丙苯和更高级的烷基苯类,在高温下也可发生侧链氯化,取代反应优先发生在α-碳原子上。
发烟硫酸表示方法:① SO3 质量百分含量 ② 游离 SO3 百分含量 w (SO3) ③ 硫酸质量百分含量 w (H2SO4) 不同表示方法之间可互换, ② 、③的互换公式如下:w (H2SO4) = 100 % + 0.225 w (SO3)w (SO3) = 4.44 [ w (H2SO4) - 100 % ]()%1001008098)()(100)(3342⨯⎥⎦⎤⎢⎣⎡⨯+-=SO W SO W SO H w例:含游离SO3 20 %的发烟硫酸换算成 H2SO4 的百分含量w (H2SO4) = 100 % + 0.225 × 20 % = 104.5 % 例:含游离SO3 65 %的发烟硫酸,试以 ① 表示5.2 芳香族磺化芳烃的磺化主要用 ①硫 酸 ②发烟硫酸 是典型的亲电取代反应。