芳香族化合物亲电取代反应
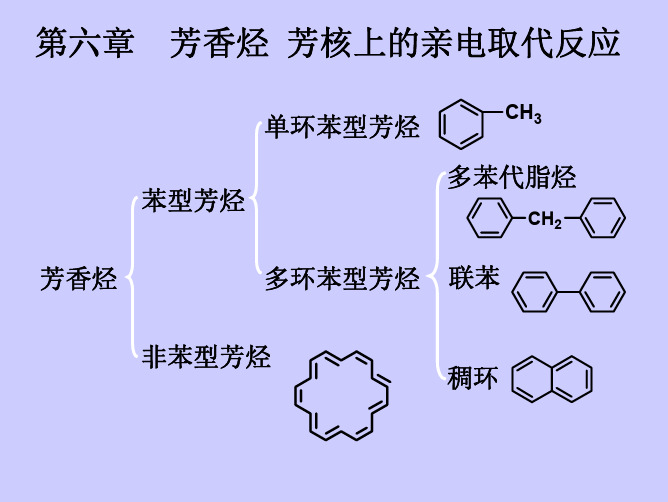
第六章 芳香烃 芳核上的亲电取代反应
AlCl3
烷基化试剂:卤代烷、烯烃或醇
Lewis酸
Lewis酸AlCl3、ZnCl2、BCl3 和无机酸 HF、H3PO4 、H2SO4
亲电试剂的形成:
R-Cl + AlCl3 → R+ + AlCl4AlCl3
70~80℃
CH(CH3)2
CH2CH2CH3
+ CH3CH2CH2Cl
机理:
(2)苯环侧链的氧化
CH3 K M nO 4 C H ( C H 3) 2
条件:α-H
COOH
COOH
氧化的特点:无论侧链多长,均被氧化成羧 氧化的特点: 基,直接连在苯环上。
CH(CH3 )2
+
COOH [O] COOH
C 2 H5
C(CH3)3
[O]
C(CH3)3
4.加成反应 加氢
+
C2H5
COOH
H
C H
C
C H
C
H
轨道杂化理论认为: 苯中碳原子sp2杂化,处在同一平面内,苯环 上所有原子都在一个平面内;键角都是120°;每 个碳原子有一个未杂化的p轨道, 6个p轨道侧面重 叠形成闭合的共轭体系。
苯形成了一个电子云密度完全 平均化了的没有单、双键之分的 大π键。苯的结构式常表示为:
H C C H C H C
CH2 CH CH2 CH3CH2 CH CH CH2 CH3 CH3
3-苯基丙烯 3–甲基-4-苯基己烷
CH=CH2
C
CH
苯基:Ph-(Phenyl的缩写) 芳基:Ar-(Aryl的缩写)
苯乙烯
3 C2H5
苯乙炔
CH3 2 1 CH3
傅克反应资料讲解

傅克反应傅-克反应傅里德-克拉夫茨反应,简称傅-克反应,是一类芳香族亲电取代反应,1877年由法国化学家查尔斯·傅里德(Friedel C)和美国化学家詹姆斯·克拉夫茨(Crafts J)共同发现。
该反应主要分为两类:烷基化反应和酰基化反应。
傅-克反应:(1)傅-克烷基化反应;(2)傅-克酰基化反应傅-克烷基化反应傅-克烷基化反应在强路易斯酸的催化下使用卤代烃对一个芳环进行烷基化。
假设使用无水氯化铁作为催化剂,在氯化铁的作用下,卤代物产生碳正离子,碳正离子进攻苯环并取代环上的氢,最后产生烷基芳香族化合物和氯化氢。
总反应式如下:傅-克烷基化机理这类反应有个严重缺点:由于烷基侧链的供电性,反应产物比起原料具有更高的亲核性,于是产物苯环上的另一个氢继续被烷基所取代,导致了过烷基化现象而形成了众多副产物。
由于这类反应是可逆的,还可能出现烷基被其他基团所取代的副产物(例如被氢取代时,也称为傅-克脱烷基化反应);另外长时间的反应也会导致基团的移位,通常是转移至空间位阻较小、热力学稳定的间位产物。
另外如果氯不是处于三级碳原子(叔碳原子)上,还有可能发生碳正离子重排反应,而这取决于碳正离子的稳定性:即三级碳>二级碳>一级碳。
空间位阻效应可以被利用于限制烷基化的数量,比如1,4-二甲氧基苯的叔丁基化反应。
1,4-二甲氧基苯的叔丁基化烷基化的底物并不局限于卤代烃类,傅-克烷基化可以使用任何的碳正离子中间体参与反应,如一些烯烃,质子酸,路易斯酸,烯酮,环氧化合物的衍生物。
如合成1-氯-2-甲基-2-苯基丙烷就可以从苯与3-氯-2-甲基丙烯进行反应:1-氯-2-甲基-2-苯基丙烷的合成曾有研究实例表明亲电试剂还能选用由烯烃和NBS生成的溴离子。
通过烯烃的傅-克烷基化在这个反应中三氟甲磺酸钐被认为在卤离子形成中活化了NBS的供卤素能力。
傅-克去烷基化反应傅-克烷基化是一个可逆反应。
在逆向傅-克反应或者称之为傅-克去烷基化反应当中烷基可以在质子或者路易斯酸的存在下去除。
亲电取代反应

亲电取代反应亲电取代反应是亲电试剂进攻化合物负电部分,取代其它基团的化学反应。
一般发生于芳香族化合物,是一种向芳香环系引入官能团的重要方法,是芳香族化合物的特性之一。
被取代的基团通常是氢原子,但其他基团被取代的情形也是存在的。
一般来说,亲电取代特指芳香亲电取代。
另一种比较少见的亲电取代反应是脂肪族的亲电取代。
中文名亲电取代反应外文名Electrophilic Substitution属性亲电取代性质反应主要反应硝化反应,卤化反应磺化反应等。
目录.1原理.2主要反应.▪硝化反应.▪卤化反应.▪磺化反应.3定位规则原理亲电取代反应主要发生在芳香体系或富电子的不饱和碳上,就本质而言均是较强亲电基团对负电子体系进攻,取代较弱亲电基团。
但对于芳香体系和脂肪体系,由于具体环境不同,其反应历程亦有所不同,现分述如下。
亲电芳香取代反应(electrophilic aromatic substitution)是芳香体系最重要的有机反应之一,常用于向芳香环系引入官能团,因此研究时间较长,在机理方面已基本达成一致。
主要反应对于亲电取代反应,其最为主要的反应类型均在芳香体系中产生,所以这里仅仅对芳香亲电取代进行一定的举例介绍。
硝化反应硝化反应苯环体系一个重要的反应,其常用于向体系引入硝基或利用硝基引入氨基等其他各种官能团,有很强的泛用性,定位选择性较好,使用最多。
由于硝基有较强氧化性,而有机体系本身又具有一定的还原性,硝基含量较多的体系就很容易成为良好的炸药材料,其中著名的TNT、苦味酸等就是通过硝化反应制备的。
Friedel(傅瑞德尔)-Crafts(克拉夫茨)反应该反应由Charles Friedel和James Crafts发现于1877年,当时采用三氯化铝等路易斯酸为催化剂,原来特指苯上的烷基化或酰基化。
但经过发展,如今泛指芳香体系中由卤代烷或酰卤为反应物在路易斯酸催化下进行的亲电取代反应。
傅克反应就反应物的不同可分为傅克-烷基化反应和傅克-酰基化反应,两者均是向芳环引入碳链的方法。
高等有机第七章+芳环上的取代反应.

7.1.3 π-络合物
HE
E+Nu- fast
E+ slow
p-络合物
HE
E
fast
+ H+
决定反应 速度步骤
动力学同位素效应可以证明此步反应速度较快
7.1.4 动力学同位素效应
用氘或氚标记苯环进行亲电取代,kH/kD或kH/kT的数值
接近1,说明C——H键断裂的步骤不是决定反应速率的
步骤。 例如:
-CF3具有强烈的-I
使苯环钝化
进攻邻位
CNFO32
H E
CNFO32
H E
CNOF23
H E
CNFO32
+ E+
对位
NCOF23
NCOF23
不稳定
CNOF32
间位
HE
CNFO32
HE
NCOF23
HE 不稳定
CNOF23
H
H
H
E
E
E
进攻邻位
NH2 H E
NH2 + E+
对位 间位
NH2 HE
NH2
加成-消除机理(Ar-SE)进行的:
HE
E
E+Nu-
k1 k-1
k2
σ-络合物
芳正离子,Wheland络合物
一般地,k2>k1,k-1,所以, σ-络合物生成步骤是决定反应 速度步骤。
7.1.1 σ-络合物存在的证明
一、分离鉴定
Me
Me Et
EtF, BF 3
- 80 oC
H
BF4
Me
Me
Me
Me
NO2+ 本位进攻生成的σ-络合物可以发生几种反应:
芳香族化合物的取代反应

(D)H (D)H NO2 H(D) HNO3/H2SO4 H(D) H(D) kH/kD = 1.05 (D)H (D)H NO2 H(D) NO2 H(D)
容易观察到较小的同位素效应 (kH/kD = 1-3,而非正常的6-7): 第一步具有可逆性及由此引起 的分配效应所产生的。
:
:
:
:OMe
+
H
E
H
E :
H
E : :OMe H E
H
E
:OMe
+
:
:OMe
+
H E
H E
+
化学
-I > +C ,钝化苯环:X
Cl
Cl E H H E
B间位定位基 的定位能力次序大致为(从强到弱) 2.
-NR3, -NO2, -CF3, -CCl3, -CN, -SO3H, -CHO, -COR,-COOH, -CONH2。
反 应 进 程
化学
2. 同位素效应 当一个反应进行时,在决定反应速率的步骤中发生 了反应物分子的同位素键的断裂,将显示初级动力 学同位素效应。最常见的是,反应物分子中的氢被 氘取代后,反应时有速率上的不同,这种变化称为 氘同位素效应,用kH/kD表示。 例如下列反应有 动力学同位素效 应,说明质子是 在决速步的失去 的:
CH2CH3 H
CH3CH2 + [AlCl3Br]
CH2CH3
H+
+
HBr AlCl3
化学
特点: 1°常用的催化剂是无水AlCl3,此外 FeCl3、BF3、 无水HF、SnCl4、ZnCl2、H3PO4、H2SO4等都有催 化作用。
亲电取代反应通式

R HCl
π-络合物
• π-络合物特点
(1)芳环的芳香性未被破坏; (2)没有与芳环形成新的化学键。
两类络合物的特点
σ-络合物
R
+HCl AlCl3
R
[ ...+... ]
HH
σ-络合物
• σ-络合物特点
(1)生成了新的σ键;
(2)芳烃的π电子体系 被破坏,形成了不稳定 的非芳香性化合物。
两类络合物的关系
芳烃与亲电试剂接触,经过π络合物, 然后形成σ络合物,即:
R +HCl
R
R
...... [ ] AlCl3
HCl
+
AlCl4-
H H (溶液)
π-络合物
σ-络合物
2.芳香族亲电取代反应的历程
经过σ络合物中间产物的两步历程
k1
H
第一步:Ar-H + E+
Ar+
k-1
E
H k2
第二步:Ar+
Ar-E + H+
E
并且生成σ络合物中间产物为控制步骤。
小结
芳香族亲电取代反应历程是经过σ络 合物中间产物的两步历程。并且生成 σ络合物中间产物为控制步骤。
1 芳香族两类络合物 2 芳香族亲电取代反应的历程
亲电取代反应
亲电取代反应通式
反应通式: R-H + Z+
R-H + Z-Y
R-Z + H+ R-Z + H-Y
1.芳香族两类络合物
芳烃+亲电试剂 [络合物] 取代产物 亲电取代反应 π-络合物 σ-络合物
两类络合物的特点
π-络合物
Rห้องสมุดไป่ตู้
+HCl
有机化学基础知识点整理芳香化合物的亲电取代反应

有机化学基础知识点整理芳香化合物的亲电取代反应有机化学基础知识点整理——芳香化合物的亲电取代反应一、引言有机化学是研究碳元素相互连结的化学性质和反应规律的学科。
而芳香化合物作为有机化学中的重要一类,其亲电取代反应具有广泛的应用价值。
本文将针对芳香化合物的亲电取代反应进行基础知识点的整理,以期帮助读者对该领域有更全面的了解。
二、芳香化合物的特点芳香化合物以脂肪族化合物为对照,其主要特点包括:1. 具有特殊的稳定性和独特的香味。
2. 芳香环中的碳原子上含有单个的π电子。
3. 和饱和化合物相比,芳香化合物更难进行化学反应。
三、亲电取代反应基本原理亲电取代反应是指一个异电子对丰富的亲电体与一个异电子对贫乏的亲核体之间的反应。
在芳香化合物中,亲电体通常是通过取代基引入的,而亲核体可以是溴离子、氨基负离子等。
亲电取代反应的机理可以分为两步:亲电体的求电子和亲核体的给电子。
四、芳香亲电体的基本性质芳香亲电体是发生亲电取代反应的关键,其基本性质包括:1. 同类反应,一般取代速率随电子效应增强而增强。
2. 不同卤原子的芳香亲电取代反应活性顺序为:I > Br > Cl > F。
3. 若取代基增加,取代反应速率常减小。
五、芳香亲核体的基本性质芳香亲核体是参与亲电取代反应的另一重要组成部分,其基本性质包括:1. 芳香亲核取代反应通常通过亲电取代碱来实现,常见的亲核取代碱包括氰离子、氧离子等。
2. 芳香亲核取代反应速率的大小与取代基的引入有关,引入的取代基越活泼,反应速率越大。
3. 芳香亲核体的选择性是通过控制反应条件和引入不同的取代基实现的。
六、芳香化合物的亲电取代反应类型芳香化合物的亲电取代反应主要包括以下几个类型:1. 麦克劳林-艾彻纳尔反应(Michaelis-Arbusov反应)2. 高夫曼反应(Hofmann反应)3. 阿群-胺裂反应(Agúndez-López反应)4. 格列巴尔反应(Gribble反应)5. 密尔斯反应(Mills反应)七、芳香化合物的亲电取代反应实例下面我们来举几个具体的芳香化合物的亲电取代反应的实例:1. 甲基苯与溴发生取代反应,得到溴代甲苯的形成。
有机化学试题芳香族化合物的取代反应

有机化学试题芳香族化合物的取代反应有机化学试题:芳香族化合物的取代反应一、引言有机化学是研究碳原子及其化合物的科学,而芳香族化合物是有机化学中重要的一类化合物。
芳香族化合物的取代反应是有机合成中的基本反应之一,它可以通过改变芳香环上的取代基,进一步合成出更复杂的有机分子。
本试题将围绕芳香族化合物的取代反应展开讨论。
二、芳香族化合物的基本结构与性质芳香族化合物是以芳香环为基础结构的有机化合物,具有特殊的化学性质。
芳香环由共轭的π电子构成,它的结构稳定且具有吸引力的电子云。
这种特殊的电子结构使得芳香族化合物具有一系列独特的物理和化学性质,如稳定性、酸碱性和亲电性等。
三、芳香族化合物的取代反应分类根据芳香环的活性和取代基的性质,芳香族化合物的取代反应可分为以下几类:1. 亲电取代反应:亲电取代反应是指在芳香环上发生的亲电性试剂与芳香族化合物发生取代反应的过程。
常见的亲电试剂包括卤代烷、酸酐和酰卤等。
亲电取代反应通常发生在芳烃的活性位点,如邻位或间位。
2. 自由基取代反应:自由基取代反应是指在芳香族化合物上发生的自由基与芳香环发生取代反应的过程。
自由基取代反应通常需要较高的反应温度或辐射条件。
常见的自由基试剂有过氧化氢和过氧化苦味酸等。
3. 核磁取代反应:核磁取代反应是指在芳香族化合物上发生的核磁试剂与芳香环发生取代反应的过程。
核磁取代反应在有机合成中具有重要的应用价值,可用于合成含特定同位素的化合物。
四、芳香族化合物的取代反应机理芳香族化合物的取代反应机理较为复杂,常见的取代反应机理有亲电取代、自由基取代和电子转移取代。
以下是亲电取代的典型机制:1. 亲电取代的机理:(1) 亲电试剂的加成:亲电试剂(如卤代烷)先与溶剂或碱性条件发生反应,生成亲电种(如卤根离子)。
(2) 芳烃的亲电攻击:生成的亲电种攻击芳香环上的活性位点,将原有的取代基脱离。
(3) 产物生成:新的取代基与芳香族化合物形成取代产物。
五、常见的芳香族化合物的取代反应举例1. 苯的取代反应:苯是最简单的芳香族化合物,常见的取代反应包括硝化反应、烷基化反应和酰基化反应等。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
亲电试剂是正离子或偶极分子中正的一端
一、 反应机理
H E E Nu
+ -
E
加成- 加成-消除机理
决定反应速度的一步是E 决定反应速度的一步是 +进攻苯 环, 果得到的是取代产物, 反应结 果得到的是取代产物, 故称为苯环上的亲电取代反应。 故称为苯环上的亲电取代反应。 缩写为SE)。 (electrophilic substitution 缩写为 )。
+ I2
I2
棕红色
它在500nm 有吸收,而苯在 有吸收, 它在 297nm处的吸收峰不复存在 处的吸收峰不复存在
σ 络合物 存在下,三甲苯和氟乙烷在- ℃ 在BF3存在下,三甲苯和氟乙烷在-80℃ 反 应,可分离出熔点为-15℃ 的中间体 可分离出熔点为 ℃
熔点为- ℃ 熔点为-15℃
二、定位效应和反应活性
三、影响邻对位比例定位效应的因素 ①空间效应 亲电试剂和苯环上原有的取代基 的体积越大,对位产物越多: 的体积越大,对位产物越多: a. 芳环上原有基团的空间效应
R
HNO3+H2SO4
R NO2 +
R
NO2ห้องสมุดไป่ตู้
R=
CH3 CH2CH3 CH(CH3)2 C(CH3)3
位 阻 依 次 增 大
含 量 依 次 减 少
N
=
O C O
=
O C N CH3
(二)定位规律的理论解释
取代基的定位效应与取代基的诱导效应、共轭效应、 取代基的定位效应与取代基的诱导效应、共轭效应、超共轭 效应等电子效应有关。 效应等电子效应有关。 诱导效应 给电子诱导效应( ),取代基上的电子通过σ键向苯环移动 ),取代基上的电子通过 苯环移动; 给电子诱导效应(+I),取代基上的电子通过σ键向苯环移动 吸电子诱导效应( ),苯环上的电子通过σ键向取代基移动。 ),苯环上的电子通过 吸电子诱导效应(-I),苯环上的电子通过σ键向取代基移动。
芳香族化合物的取代反应
§1、芳环上的亲电取代反应 、
苯环的离 域π 轨道
芳环上离域的π电子的屏蔽作用,易于发生亲电取代反应 芳环上离域的 电子的屏蔽作用,易于发生亲电取代反应; 电子的屏蔽作用 只有当芳环上引入了强吸电子基团,才能发生亲核取代反应。 只有当芳环上引入了强吸电子基团,才能发生亲核取代反应。
亲电取代主要得到邻对位产物, 亲电取代主要得到邻对位产物,可以从反应中间体碳正 离子的极限式来分析,以甲苯为例: 离子的极限式来分析,以甲苯为例:
CH3 进 攻 邻位 : + E
+ +
CH3 H E
+
CH3 H E
+
CH3 H E
CH3 进攻对位: + E
+ +
CH3
+
CH3
① CH3
+
H
CH3 进 攻 间 位: + E
如:―OH、―OR、―NR2、―OCOR 等。 、 、
以苯酚为例: 以苯酚为例
HO +C> I
进 攻 邻位 :
OH
+
OH H E
+
OH H E
+
+OH
H E
③
+
H E OH
OH 进 攻 对位 :
+
OH
+
OH
+
H 进 攻 间位 :
E OH
+
H
E OH
H
E OH
H
④
E
H E
+
H E
+
H E
③④两种极限 ③④两种极限 结式每个原子 都有完整的外 电子层结构。 电子层结构。
这类定位基可使苯环活化(卤素除外) 其特点为: 这类定位基可使苯环活化(卤素除外)。其特点为: a. 带负电荷的离子。如: 带负电荷的离子。
O
b. 与苯环直接相连的原子大多数都有未共用电子对, 与苯环直接相连的原子大多数都有未共用电子对, 且以单键与其它原子相连。如: 且以单键与其它原子相连。
CH3 N O X N CH3 O O C
CH3 CH3 CH3 + R
R X AlCl 3
R
R=
CH3 CH2CH3 CH(CH3)2 C(CH3)3
位 阻 依 次 增 大
依 次 减 少
依 次 增 多
结论:对位产物随新引入基团体积的增大而增多。 对位产物随新引入基团体积的增大而增多。
如果芳环上原有基团与新引入基团的空间位阻都很大 时,对位产物几乎为100%。 对位产物几乎为 。
c. 与苯环直接相连的基团可与苯环的大π键发生σ,π–超 超 共轭效应或具有碳碳重键 或具有碳碳重键。 共轭效应或具有碳碳重键。如:
CH3 C6H5 CH =CH2
=
CH3
2. 第二类定位基(即间位定位基) 第二类定位基(即间位定位基) ―N+(CH3)3 > ―NO2 > ―CN > ―SO3H > ―CHO > ―COOH > ―COOR > ―CONH2 >―+NH3 等. 这类定位基它们使苯环钝化。其特点是: 这类定位基它们使苯环钝化。其特点是: a. 带正电荷的正离子。如: ―N+(CH3)3 、 ―+NH3 。 带正电荷的正离子。 b.与苯环直接相连的原子以重键与其它原子相连, b.与苯环直接相连的原子以重键与其它原子相连,且重键 与苯环直接相连的原子以重键与其它原子相连 末端通常为电负性较强的原子。 末端通常为电负性较强的原子。如:
共轭效应 给电子共轭效应( ),取代基的P电子向苯环迁移 ),取代基的 电子向苯环迁移; 给电子共轭效应(+C),取代基的 电子向苯环迁移; 吸电子共轭效应( ),苯环的π电子向取代基迁移。 ),苯环的 吸电子共轭效应(-C),苯环的π电子向取代基迁移。 多数取代基既可共轭, 多数取代基既可共轭,也可诱导 。大部分共轭和诱导的方 向是一致的。特例:卤素( , ) 向是一致的。特例:卤素(-I,+C) 。
π络合物 络合物
σ 络合物
一般来说, 络合物的形成是可逆的 络合物的形成是可逆的; 一般来说, π络合物的形成是可逆的; 络合物的形成基本上是不可逆的, σ 络合物的形成基本上是不可逆的, 且通常是速度的控制步骤。 且通常是速度的控制步骤。
络合物是通过电荷转移形成的, π 络合物是通过电荷转移形成的,因此也 叫电荷转移络合物。 叫电荷转移络合物。
+
E
CH3
+
H
E
H
E
②
CH3 H E
+
CH3 H E
+
H E
效应的基团,它又可分为: (2)具有 和+C 效应的基团,它又可分为: )具有–I
A. +C >–I 的基团,综合影响的结果:苯环的电子 的基团,综合影响的结果: 云密度增加,使苯环活化。取代基进入邻、对位。 云密度增加,使苯环活化。取代基进入邻、对位。
以氯苯为例
Cl
+
进攻 邻 位:
Cl H E
+
Cl H E
+
+ Cl
H E
⑦
+
H E Cl
⑤
Cl 进攻 对 位:
+
Cl
+
Cl
+
H Cl 进攻 间 位:
E
H
⑥
E
H Cl H E
E
H⑧ E
Cl
+
H E
+
+
H E
每个原子都有完整 的外电子层结构。
2. 第二类定位基(即间位定位基) 第二类定位基(即间位定位基)
效应的基团, (1)表现为 、+C效应的基团,但这里的 效应是通 )表现为+I、 效应的基团 但这里的+C效应是通 超共轭效应使苯环致活的。 过σ,π–超共轭效应使苯环致活的。如: 超共轭效应使苯环致活的 ―CH3、―CH2X (X=F、Cl、Br、I)。 = 、 、 、 。
致活原因(以甲苯为例):甲基具有给电子的电子效应, 致活原因(以甲苯为例):甲基具有给电子的电子效应,使苯 ):甲基具有给电子的电子效应 环的电子云密度增加,一方面使亲电试剂更容易进攻苯环, 环的电子云密度增加,一方面使亲电试剂更容易进攻苯环,同 时也使反应过程中产生的碳正离子的电荷得以分散, 时也使反应过程中产生的碳正离子的电荷得以分散,所以更容 易被取代。但由于电子效应是微弱的, 易被取代。但由于电子效应是微弱的,所以对苯环的活泼性影 响也较弱。 响也较弱。
愓
类别 性质 取
O NR2 NHC R NHR O OC R
CH3 CR3
代
O
NH2 OH OR
F Cl Br
I
基
电子 +I 效应 +C
NO2 CN SO3H CHO COCH3 COON COOCH3 CONH2
NR3
+C>-I >
+I
+C< < -I
-C -I
-I
1. 第一类定位基(即邻对位定位基) 第一类定位基(即邻对位定位基)
(CH3)3C (Cl、Br) 、 浓 H2SO4 (CH3)3C (Cl、Br) 、 SO3H ~100%
B.+ <–I 的基团,综合影响的结果:使苯环 .+C 的基团,综合影响的结果: .+ 钝化,取代基进入邻、对位: 钝化,取代基进入邻、对位:
X I > +C
至钝原因(以氯苯为例): 至钝原因(以氯苯为例): 苯环上电子云密度降低: +C <–I ,苯环上电子云密度降低:亲电试 剂不易进攻苯环; 剂不易进攻苯环;使中间体碳正离子更不稳 反应时过渡态势能增大。 定,反应时过渡态势能增大。