假肥大型肌营养不良症一家系报告分析
假性肥大性肌营养不良

心理状况评价
了解患儿的心理状况,如情绪、 社交能力、自尊心等。
社会参与评价
评估患儿在社会和家庭中的角色 和参与程度,以及其对生活质量
的影响。
04 诊断方法与标准
病史采集和家族史分析
01
详细了解患者的起病年龄、病程 、症状及进展情况。
02
询问家族史,特别注意家族中是 否有类似病例或遗传性疾病史。
体格检查及辅助检查
分型
根据临床表现和遗传特点,假性肥大性肌营养不良可分为杜氏肌营养不良(DMD)和贝克肌营养不良(BMD) 两种类型。DMD病情较重,进展较快,患者多在青少年期失去行走能力;BMD病情相对较轻,进展较慢,患者 可能保持较长时间的行走能力。
诊断标准与鉴别诊断
诊断标准
结合患者的临床表现、家族史、血清学检查和肌肉活检等结果进行综合判断。 其中,肌肉活检发现肌肉细胞中的抗肌萎缩蛋白缺失或功能异常是确诊的关键 依据。
吞咽困难
心理问题
部分患者可能出现吞咽困难,需要进行吞 咽功能训练,必要时给予鼻饲或胃造瘘术 。
由于疾病影响,患者可能出现自卑、抑郁等 心理问题。应给予心理支持和辅导,帮助患 者建立积极的生活态度。
07 患者教育与家庭护理
患者教育内容
疾病知识普及
向患者和家属详细解释假性肥大性肌营养不良的病因、发病机制、 临床表现、诊断方法和治疗方案。
针灸治疗
通过刺激穴位,调和气血,达到治疗目的。
3
基因治疗
针对基因突变进行干预,目前仍处于研究阶段。
康复训练与心理支持
运动疗法
根据患者病情制定个性化运动方案,如肌力训练、关节活动度训练 等。
心理干预
提供心理咨询和支持,帮助患者和家属应对情绪困扰,提高生活质 量。
女性假肥大型肌营养不良症

女性假肥大型肌营养不良症*导读:本文介绍女性假肥大型肌营养不良症的发病机制等相关知识。
……1.女性DMD的诊断目前尚无假肥大型肌营养不良症(DMD)女性患者的独立诊断标准。
DMD的诊断主要根据发病年龄、临床表现、遗传方式、基因检测和抗肌萎缩蛋白检测,配合肌电图、肌肉病理检查和血清肌酶的测定,以及心电图及心脏彩色超声。
本研究两位患者均幼年起病,出现进行性四肢近端肌肉萎缩、无力,双腓肠肌假性肥大,肌电图呈肌源性损害,血清肌酶水平显著增高, 肌肉病理结果符合DMD的表现,男患者抗肌萎缩蛋白(-),女患者抗肌萎缩蛋白免疫组织化学染色可见少量肌纤维有不连续膜阳性,其中女患者有窦性心律不齐, DMD诊断成立。
尽管基因检测未发现致病突变,但是结合肌肉组织抗肌萎缩蛋白免疫组织化学及生化、电生理等结果可以作出DMD的诊断。
女性DMD有时需与某些常染色体隐性遗传的肢带型肌营养不良症( lmi b girdle muscular dystrophy, LGMD)相鉴别,其中LGMD2C、2D、2E、2F型在临床上类似于DMD,曾被称为重型儿童常染色体隐性遗传性肌营养不良。
这几种类型的LGMD患儿开始走路通常较同龄儿晚, 4~10岁发病,表现为走路易跌,上楼困难,上肢上举无力,伴有腓肠肌肥大、心肌病变和疾病早期血清CK水平显著增高,智力正常。
大多数患者在9~16岁丧失行走能力,通常在9~19 岁死亡。
该病与DMD最明显的区别在于:常染色体隐性遗传;肌膜免疫组织化学染色表现为α-肌葡聚糖缺乏,而抗肌萎缩蛋白保留,据此可与本例患病兄妹相鉴别。
多重PCR是用于检测抗肌萎缩蛋白基因的常用方法,但不能用于携带者的筛选;连锁分析虽然可以用于携带者的筛选却受制于样本量的大小。
MLPA 是新近创建的,可以准确有效检测抗肌萎缩蛋白基因中较大的缺失和重复突变的实验方法。
MLPA 用于抗肌萎缩蛋白基因检测的优势在于高效、高灵敏度,更重要的是可用于携带者的基因检测。
Duchenne型肌营养不良症家族遗传学分析

Duchenne型肌营养不良症家族遗传学分析*导读:本文介绍Duchenne型肌营养不良症家族遗传学分析。
……Duchenne型肌营养不良症(DMD),即假肥大型进行性肌营养不良症,是一种X连锁隐性遗传的致死性神经肌肉疾病,以男性受累为主,发生率占出生男婴的1/3 500左右,平均确诊年龄4·6岁,以骨盆和肩胛带肌以及股四头肌无力、萎缩并伴有腓肠肌假性肥大为特征,病程进展较快,大多在20岁左右因心肺衰竭而死亡。
目前本病没有很好的治疗方法,因此产前筛查和携带者的检出及正确的遗传咨询尤为重要。
该家族的致病基因来自于系谱图中的Ⅰ2,Ⅰ2、Ⅱ3、Ⅲ2、Ⅲ4、Ⅳ10均为表型正常的女性,因生出患病的儿子,而证明其均为肯定的DMD致病基因携带者;Ⅱ4、Ⅲ3尽管表型正常,也无生出患病的儿子,但理论上仍有1/2的可能性是携带者。
Ⅲ6的情况比较特殊,按孟德尔理论分析,她有1/4的可能性为致病基因的携带者,但由于她生育的5个儿子都正常, 因此按Bayes定理分析,她只有1/97的可能性为携带者,风险大大降低。
系谱中先证者Ⅴ1之母Ⅳ10 现又怀孕,已诊断出胎儿性别为男性,那么此男性胎儿Ⅴ2患病的可能性为1/2,风险很高,后做产前 DMD基因PCR诊断,证实其为患者,已引产。
若还准备再次生育,建议选择生育女孩。
由于第四代中的正常个体均已进入生育年龄,他们今后的生育情况需做遗传分析:①家系中只要超过发病年龄且表型正常的男性,其基因型也一定正常。
②第4代中的8个男性以及第4代的女性Ⅳ1,现已进入生育年龄,只要他们的配偶正常且家系中无DMD的家族史,后代的发病风险等同于普通人群;③只有Ⅳ2是一个高风险的个体,根据孟德尔定理有1/2的可能性携带有致病基因,怀孕后可做胎儿基因的产前诊断,或选择生育女孩。
除上述的系谱分析法判断携带者之外,也可测定血清CPK活性予以佐证。
系谱分析中为肯定携带者和疑似携带者的女性中, CPK 异常占80% ~90%。
面肩脓型进行性肌营养不良症一例大家系调查报告1)
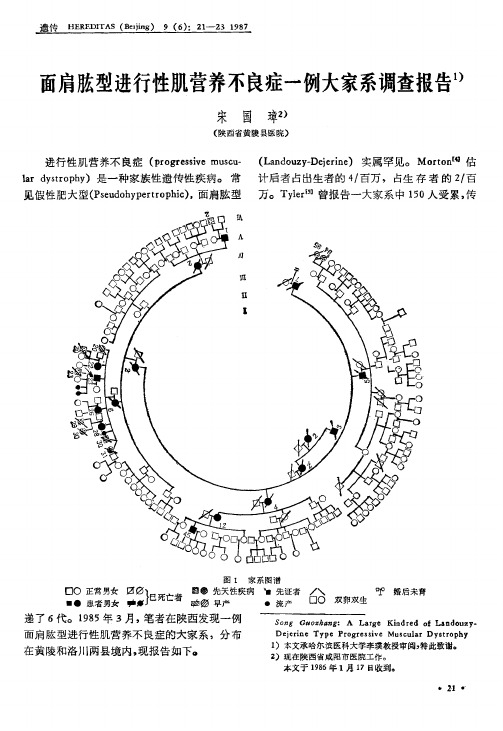
V -V I代之间消灭该病致病性基因的 传递 , I I
‘ 正面; b .背面.
. 22 ,
至 VI I代完全杜绝遗传病儿出生, 从此终止本
病的传递。
参 考 文 献
出版社 , 12 第 3 页。 E 肖 3〕 永义:18。大众医学, 5: 95 () 20 5
[礴]
M r n N E: 9 A e. H m G nt 1: ot , . 15. r J u . e , o . 9 m . e . 1
2 V I , ( I 9系开放性神经管畸形,并右前臂缺如, 一个家系而出现的偏倚;() V 代中出现 V- 2 V-0 I3 系先天性肌营养不良) 其姨母 ( -2 多种先夭性疾病 ( -4 V 9 。 I 1) V V 9, 5患白化症, - - V 1 V- , 2系多指畸形,V- 0 V -3 VI2, I I2, 2 , -9 I 和姨表兄 ( -, 4)均已丧失劳动力。 V1 V 5 -
见假性肥大型(suoyet pi , Pedhpr oh ) 面肩脑型 r c 万。T l [ yr e”曾报告一大家系中 10 5 人受累, 传
图 1 家系图潜 口O 正常 男女
二
翻哪 先天性疾病 、 先证者 Z 吵 、 已死亡者 口 O 双卵双生 患 者男女 叻娜 叻扬 早产 方 法
本文调查对象为上海市利群医院高血压专 q / p为2 8 .0 6 科门诊患者, 根据符合上述诊断标准的先证者, ( 三)子女
除详细询问病史外, 并作胸透、 眼底、 尿常规、 心
电图等检查。 确诊为原发性高血压病, 排除了 继发性高血压的可能。 对家属调查采取询问与检 查 相 结 合 的方
时, 子代都不发病;()连续发病, 4 无隔代遗传 现象 ( I代尚未到发病年龄, V 目前亦无病象) , 符合常染色体显性遗传。 该家系内 发病 率 为 1八3 , 6 7 无近亲婚配。 因传代久远,对于致病 基因的起源, 是由 I2本身的基因突变所引起, - 抑或由其上代传递而来, 已难确定。 值得注意的是:()该家系内仅见单纯的 1 母系遗传。父系竟无一人传递本病,这一现象 应属其固有的遗传特征,抑或因调查仅局限于
专家述评:《中国假肥大型肌营养不良症诊治指南》解读

专家述评:《中国假肥大型肌营养不良症诊治指南》解读中华医学会神经病学分会神经肌肉病学组以及肌电图与临床神经生理学组联合编写的《中国假肥大型肌营养不良症诊治指南》(以下简称指南)于2016年发表在《中华神经科杂志》后,收到全国基层医院和专家学者各方面的反馈信息,对临床工作有较好的指导作用。
为了能够更好地理解指南并应用于临床实践,笔者根据各方面反馈信息对指南进行解读。
一、撰写背景Duchenne型肌营养不良症(DMD)亦称假肥大型肌营养不良症,系DMD基因突变导致的X连锁隐性遗传性肌肉病,发病率约1/3500活产男婴。
据此推算,我国有(70-90)×10^3例Duchenne型肌营养不良症患者,遍布全国各地区,无地域特征,平均每省不足3000例。
尽管该病具有明显的临床特征如小腿腓肠肌肥大、Gowers征阳性、血清肌酸激酶(CK)显著升高,但临床少见,因此部分医师不熟悉其诊断、治疗和预防。
近年来,Duchenne型肌营养不良症研究取得突破性进展,新的治疗方法涌现,因此,中华医学会神经病学分会组织编写指南,以指导Duchenne型肌营养不良症的诊断、治疗和预防。
二、指南框架经中华医学会神经病学分会指南起草专家委员会相关专家多次讨论,最终确定从概述、诊断、治疗、预防四方面撰写,重点是诊断与治疗,详细描述诊断过程中的临床表现、辅助检查和鉴别诊断以及常规治疗方法,具有可操作性;对于新的治疗进展如基因治疗和干细胞移植治疗,尚处于临床研究阶段,简要介绍,但附有重要参考文献,有助于读者了解疾病治疗发展方向,有兴趣的读者可以自行查阅相关参考文献。
1. “概述”主要介绍Duchenne型肌营养不良症的定义和发病率由于Duchenne型肌营养不良症病因明确,故定义突出该病是DMD基因突变导致的X连锁隐性遗传性肌肉病,以及特征性临床表现、预后和可能的发病机制。
近年关于Duchenne型肌营养不良症发病率的文献报道较多,我们仍采用流行病学设计合理、调查全面的经典数据,发病率为1/3500活产男婴。
肌营养不良症疾病研究报告

肌营养不良症疾病研究报告疾病别名:肌营养不良症所属部位:全身就诊科室:神经内科,脑外科病症体征:不能露齿及突唇,肌肉肥大,肌肉萎缩,无力,眼睑下垂,易跌倒疾病介绍:什么是肌营养不良症?肌营养不良症按照字面意思是指消瘦或者肌肉萎缩,肌营养不良症是一组以进行性加重的肌无力和支配运动的肌肉变性为特征的遗传性疾病群,肌营养不良症有很多种类型,有些是在出生时就可以观察到的先天性肌营养不良症,而其他的则是在青春期才观察到的,不考虑发病的确切时间,有些肌营养不良症会导致运动受损甚至瘫痪症状体征:肌营养不良症有什么症状?(一)假肥大型呈性环连隐性遗传,男性罹病,女性携带。
通常在幼儿期起病。
表现为能走路的年龄推迟,行走缓慢、易跌,跌倒后不易爬起。
多数有小腿肌的肥大,病初肥大肌肌力相对稍强。
臀中肌受累而致骨盆左右上下摇动;跟腱挛缩而足跟不能着地;腰大肌受累而腹部前凸,脑后仰。
呈鸭型步态。
从蹲位只有靠两手撑着自己身体而逐步站直大腿,然后逐步挺起身子。
继骨盆带肌肉受累之后,逐步出现肩胛带肌肉萎缩、无力,两臂举高不能。
菱形肌、前锯肌、肩胛肌、岗上、岗下肌萎缩而使肩胛游离、肩胛骨呈翼状耸起,称翼状肩。
病程逐步发展,某些儿童可能由于本身生长发育的影响,出现病程的相对稳定或好转。
多数病孩在10岁时已丧失行走能力,依靠轮椅或坐卧不起,出现脊柱和肢体畸形。
晚期,四肢挛缩,活动完全不能。
常因伴发肺部感染、褥疮等于20岁之前丧生。
智商常有不同程度减退。
半数以上可伴心脏损害,心电图异常。
早期呈现心肌肥大,除心悸外一般无症状。
(二)面一肩一肱型呈常染色体显性遗传。
男女均可罹病。
病情严重程度不一,轻者可无任何主诉,在偶然机会或医师进行家谱分析时发现。
幼年或青春期隐匿起病,常在发病后数年才被引起注意。
面肌受累较早,表现为睡眠时闭眼不紧、吹气无力、苦笑脸容。
逐步出现颈肌、肩胛带肌、肱肌的萎缩、无力。
肩胛带和肱部肌肉萎缩,两侧肩峰隆突明显。
整个肩胛部酷似衣架。
假性肥大性肌营养不良解读
假肥大型肌营养不良是由遗传因素所致的以进行性骨骼肌无力为特征的一组原发性骨骼肌坏死性疾病, 临床上主要表现为不同程度和分布的进行性加重的骨骼肌萎缩和无力。
可累及心肌。
是小儿时期最常见的遗传性肌病,今天我们主要从以下几个方面进行了解 ,一、分类1,['bekə]Becker 型进行性肌营养不良 :或仅轻度受累, 预后较好,又称良性型。
2,Duchenne(du:'ʃen 型肌营养不良二、病因假肥大性肌营养不良中,有约 1/3病例是由新发生的基因突变引起, 即父母双亲中, DMD 基因都正常, 但个体胚胎发育时, 出现了 DMD 基因的突变, 导致疾病的发生。
这种突变发生的个体,可将致病基因遗传给后代。
三、发病机制:与肌营养不良发病有关的膜结构蛋白是由多种蛋白质组成的一个大复合物称抗肌萎缩蛋白 -糖蛋白复合物 (DGCdystrophin[dis'trɔfin]的一端与肌动蛋白相联结, 另一端与β-dystroglycan 相联结再通过α-dystroglycan 与基底膜上的细胞外基质蛋白α2-Laminin 相联结在结构上起到了连接肌肉细胞内肌动蛋白和细胞外基质的桥梁作用。
疾病晚期,几乎所有肌纤维已经变性时,血清 CK 含量反可下降。
HE 染色×200 ATP染色×200四、临床表现 ['ɡau ə]鸭步 :患儿出生时或婴儿早期运动发育基本正常,少数有轻度运动发育延迟,或独立行走后步态不稳, 易跌倒。
一般 5岁后症状开始明显, 髋带肌无力日益严重, 行走摇摆如鸭步态, 跌倒更频繁, 不能上楼和跳跃, 肩带和全身棘力随之进行性减退, 大多数 10岁后丧失独立行走能力 20岁前大多出现咽喉肌肉和呼吸肌无力,声音低微,吞咽和呼吸困难,很易发生吸入性肺炎等继发感染死亡, BMD 症状较轻,可能存活至40岁左右。
Gower 征 :由于髋带肌肉早期无力,一般 3岁后患儿即不能从仰卧位直接站起,必须先翻身成俯卧位, 然后两脚分开, 双手先支撑于地面, 继而一只手支撑到同侧小腿, 并与另一手交替移位支撑于膝部和大腿上,使驱赶从深入鞠躬位逐渐竖直,最后成腰部凸的站立姿势。
可治性罕见病—假肥大型肌营养不良症
可治性罕见病—假肥大型肌营养不良症一、疾病概述假肥大型肌营养不良症( pseudohypertrophic muscular dystrophy),是一种严重的神经肌肉疾病,是进行性肌营养不良中最常见,也是小儿时期最常见、最严重的一型[1]。
多见于学龄前和学龄前期,分为杜氏肌营养不良( Duchenne muscular dystrophy.DMD)和贝克肌营养不良(Becker muscular dystrophy,BMD),其中DMD 病情严重。
DMD发病率为1/6 291~1/3 802活产男婴[2],BMD仅为其1/10。
研究发现两者均是由于抗肌萎缩蛋白基因(dystrophin基因,亦称DMD基因)突变所致[3],其主要的突变类型为基因的部分缺失或重复,占全部突变基因的50%~70%,引起肌纤维膜的稳定性下降,从而导致肌纤维不同程度的坏死、变性和再生,伴随肌纤维肥大、萎缩和间质的结缔组织增生,一般没有炎症细胞增生的病理改变。
属X-连锁隐性遗传病,一般是男性患病,女性携带突变基因。
然而实际上,仅2/3患者的病变基因来自母亲,另1/3患者是自身抗肌萎缩蛋白基因的突变,此类患儿的母亲不携带该突变基因,与患儿的发病无关。
抗肌萎缩蛋白位于肌细胞膜脂质中,对稳定细胞膜,防止细胞坏死、自溶起重要作用。
定量分析表明,DMD患者肌细胞内抗肌萎缩几乎完全缺失,故临床症状严重,而抗肌萎缩蛋白数量减少则导致BMD,后者预后相对良好,病程进展相对缓慢。
由于该蛋白也部分地存在于心肌、脑细胞和周围神经结构中,故部分患者可合并心肌病变、智力低下或周围神经传导功能障碍。
二、临床特征主要表现包括:(1)缓慢进行性加重的对称性肌无力、肌萎缩和运动功能倒退,一般5岁后症状开始明显,大多数13岁丧失独立行走能力,20岁左右出现咽喉肌肉和呼吸肌无力,声音低微,吞咽和呼吸困难,最后因呼吸衰竭或心力衰竭而死亡;BMD 病情变化多样,进展相对缓慢,在首次发病15~20年后仍可有行走能力。
假肥大型进行性肌营养不良1例家系调查
家庭成员进行详细询问, 绘出系谱图( 见附图) 。
从本例系谱图可看 出患者都是男性 , 双亲无 病, 女儿不发病 , 是肯定致病基 因携带者。Ⅲ 但 的父 亲 Ⅱ 和 叔 父 Ⅱ。 患 者 ,。I 型均 正 常 , 是 I、 表
第 2 2医院优 生优育研 究中心 0
闭经 的可能性 , 有 利 于 疾 病 的 治疗 以及 闭 经 系 且
中有 8例 出现 不规 则 流血 , 1例超 过 6月 , 能 有 可
与 L G—I S对子 宫 内膜 血 管 影 响 有关 。L G— N U N I S可 使 子宫 内膜 血管 扩 张 , U 管壁 变 薄 , 管 周 围 血 支持 组 织减 少 , 同时 引起 子 宫 内膜 多 种 血 管 生 长
本组 2 9例 子 宫 和 宫 腔 均 有 不 同 程 度 增 大 ( 腔 8 —1 e , 例 脱 环 者 子 宫 腔 均 超 过 官 3 m) 2
1丰有 吉 .妇产科学. 北京 : 民卫生 出版社 ,0 2 3 8— 人 20 : 4
3 9 4
1c 0 m。为处理脱环我们建议 : 向患者讲清脱环 ①
为 x连锁隐性遗传病 , 是一组 由遗传因素引起 的
原发 性 骨骼 肌疾病 。临床 主要表 现为缓 慢进 行 的 肌 肉萎 缩 、 肉 无 力 及 不 同 程 度 的 运 动 障 碍 。 肌
院血 清 酶 测定 、 检测 、 电 图及 活 检 等 检查 , 尿 肌 确 诊 为 进行 性肌 营养 不 良症 。其 叔 父 Ⅱ 一子 Ⅲ ( ,
中西 医结合 治疗 , 如卡 洛环 钠 +独一 味 、 例 葆宫 止
宫 , 良反应 较轻 的优 势 , 不 为治疗 子宫 腺肌症 提供 了一个 方便 、 经济 、 有效 的保 守治 疗途 径 。但 是在
假肥大型肌营养不良15例临床分析
可延缓病情发展 。基 因治疗近几年来取得 了很大的进展 , 给
山东 医药 2 0 0 8年第 4 第 4 8卷 7
力疝修 补术 用补 片修 补 缺损 部 位 , 使用 的补 片 具 有
良好 的组织 相容性 和抗感染 性 , 能分散 腹壁 张力 , 迅
T R 可 以在 T R u P, u P完 成 后 , 快 结束 手 术 。本 组 尽 l 均先行腹 股 沟疝修 补 , 行 T R 无严 重并 发 6例 再 u P,
症 出现 。
速与 组织 固定牢 靠 。术 中先 游 离 出疝 囊 , 高 位结 行 扎及缝 扎 。直疝如果 疝囊 较小 , 可不结 扎疝囊 , 直接
术前 应 充分 评 估 患者 的基本 情况 , 积极控 制 各 种合 并症 。对 于体质 差 、 不能耐受 一期手 术 的患 者 , 选 择二期 手术 更 为合 适 。对 于 有 手 术 禁 忌 证 的 患 者, 应延期 手术 。术后 应 积极预 防感染 , 制膀胱 痉 控 挛, 减少便 秘 、 咳嗽等 可致腹 压增 大的 因素 。 [ 参考 文献 ]
发病年龄较晚 ( 5—2 在 0岁发 病) 发病前 可能 已存在 假性 ,
肥大 , 往往 不 能 引起 注意 。病 情进 展 速度 慢 ( 程 可达 但 病 2 以上 , 5a 往往 2 0岁 以后 仍能行走 ) 多不伴有 心肌受 累或 , 仅轻度受 累, 预后较好 , 又称 良性型。 本组 l 5例均为杜 氏进行性肌营养不 良。积极 的对症 和 支持治疗有助于提高患儿生活质量及延长生命 , 包括鼓励并
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
1% , 本 病 的 一 种 重 要 的 突 变 形 式 _ 。分 子 生 物 学 技 术 0 是 2 J
完成 了该 病 的 基 因 定 位 、D A克 隆 、 显 子 全 序 测 定 等 关 键 eN 外
目前 对 该 病 的 治 疗 没 有 特 殊 疗 法, M 患 者 中 1 D D / 3是 新
生 突 变 ,/ 23由 遗 传 而 来 , 以及 时 对 携 带 者 进 行 检 测 有 着 十 所
分重 要 的 临床 意义 。截 止 到 目前 最 有 希 望 的 疗 法 就 是 利 用 反
吉林医学 21 02年 8月 第 3 3卷第 2 期 3
义 寡 核 苷 酸 ( 0 s诱 导外 显 子 跳 跃 , 修 复 dsohn 录 本 A N) 以 ytpi 转 r
的 读 码 框 。 预 防 本 病 的最 有 效 的措 施 就 是 产 前 检 查 , 外 对 另 患 者 进 行 精 细 的 致 病 基 因检 测 和遗 传 学 诊 断 , 合 支 持 治 疗 , 配
吉 林 医学 2 1 0 2年 8月 第 3 3卷第 2 期 3
・
51 7 ・ 4
胞受损及粘连性肠梗阻等并发症 。
质子治疗是在放 疗基 础上发展 出来 的肿瘤 治疗 技术 , 具 有 能 量 大 , 透 力 强 , 经 的 正 常 组 织 损 伤 小 和 治 疗 时 间 短 等 穿 途
走、 动、 活 劳动 能 力 均 正 常 。查 体 : 质 消 瘦 , 育 一 般 , 肢 体 发 四
肌力 , 张 力 正 常 , 见 肌 肉 假 性 肥 大 。 肌 酶 检 查 ; O 、 肌 未 C T
1 家 系 分 布
H D C L H 均在 正 常 , 色 体 4 x ( / ) B H、 K、 D 染 6 y G G G带 显 示 染 色 体
涎 腺 淋 巴 上皮 癌 ( m h p hl a i m E )他 的发 1 po i eal r n aL C , y et i e eo 生 目前 认 为 是 涎腺 的小 管 一腺 泡 复 合 体 或 导 管 在 致 瘤 因 子 作 用 下 增 殖 而成 。在 涎 腺 淋 巴上 皮 癌 的发 生 过 程 中 E B病 毒 是
染 色 体 核 型 4 x( / ) 6 x G G G带显 示 患 者 染 色体 组 型 分析 正 常 。
者 通 过 先 证 者 线 索 追踪 一 家 系 D D的 病 情 进 展 和 变 化 , 将 M 现
具体 情况报告如下。
Ⅱ8为 患 J 4 舅 , 患 儿 来 就诊 , 时 没 有 不 适 的 感 觉 , L, 陪 平 行
丘 王 陈 宁 (. 南 大 学 湘雅 医学 院 , 南 长 沙 f, j 1中 湖 400 ; . 南 省 临 沧 市 人 民 医院 , 南 临 沧 1082 云 云 670 ) 700
[ 关键 词 】 假 肥 大 型 肌 营养 不 良症 ; 系 ; 析 家 分
进行 性 肌 营 养 不 良症 的 发 生 与 遗 传 因 素 存 在 一 定 的 联 系 , 床 主 要 表 现 为 肌 肉 萎 缩 和 无 力 。假 肥 大 型 肌 营 养 不 良 临 症( M ) 进行性 肌营养 不 良症的一 种临床 常见 的类 型 , D D为 笔
展 加 重 失 去独 立行 走 能 力 。
经 济 的 条 件 没 有 及 时 得 到 治 疗 , 情 呈 进 展 性 加 重 , 终 2 病 最 2 岁时死于肺部感染 。 l I 患 儿 母 亲 , 时 没 有 不 适 的 感 觉 , 走 、 动 、 动 3为 平 行 活 劳 能力 均 正 常 。 查 体 : 清 , 育 正 常 , 养 可 , 特 殊 面 容 , 神 发 营 无 心
情稳定 , 在随访之中。 尚
特 点 , 合 于 全 身 肿瘤 的治 疗 。但 质 子 技 术 也 有 其 局 限 性 : 适 相
对 固定 , 限 的 器 官 肿 瘤 如 脑 、 、 宫 、 列 腺 等 疗 效 较 好 ; 局 肝 子 前
活 动 的 , 漫 的 器 官 肿瘤 如 胃 、 肠 等 则 疗 效 较 差 。质 子 技 术 弥 小 治 疗 间 皮 瘤 尚未 见 正 式 报 道 , 由 于成 年 人 以腹 式 呼 吸 为 主 , 但 腹 膜 是 随着 呼吸 起 伏 运 动 的 , 合 本 例 肿 瘤 又 比 较 弥 漫 , 及 结 涉 到 了肝 、 层 腹 膜 、 肌 下 缘 等 处 , 脏 膈 因此 个 人 认 为 不 太 适 合 。
4 小 结
患 者 因进 行 性 腹 胀 , 水 1 院 , 腹 腔 镜 确 诊 为 弥 漫 腹 2d入 经 型 腹 膜 恶 性 问皮 瘤 。 由于 患 者 不 同 意 全 身 化 疗 , 次 给 予 顺 首 铂 腹 腔 化 疗 后 腹 水 完 全 消 退 。 目前 已 行 第 七 次 局 部 化 疗 , 病
工 作 。 在 D D基 因 突 变 中 , 有 3 % 是 新 发 突 变 , 他 突 变 M 约 0 其
以 X一连 锁 隐 形 遗 传 , 以 此 病 主 要 好 发 于 男 性 0 。 由 于 基 所 】
因 突 变影 可 引发 一 系 列 酶 及 实 验 室 指 标 的 改 变 , 得 近 端 骨 使 骼肌 呈 进 行 性 萎 缩 、 力 和 腓 肠 肌 假 性 肥 大 状 态 , 着 病 情 进 无 随
时年 龄 小 , 处 于 儿 童 期 ; 起 病 较 隐 匿 , 易察 觉 , 发 症 状 正 ② 不 首
图 1 家 系 谱 图
Ⅱ 1
通常 会 表 现 为 骨 盆 带 肌 无 力 、 张 力 减 低 、 走 呈 鸭 步 、 易 肌 行 容
跌倒 等行为 ; 随着病情 的进一 步发展 , 端肌 肉有所萎 缩 , ③ 近 腓 肠 肌 假 性 肥 大 已呈 现 出 来 ; 伴 有 反 复 无 常 的 呼 吸 道 感 ④ 染 ; 辅 助 检 查 : 证 者 肌 肉酶 学 检 查 明 显 高 于 正 常 ; 先 证 ⑤ 先 ⑥ 者 的母 亲 肌 肉酶 学 检 查 C 、 D 、 B H 均 高 于 正 常 。 KLHH D
肺检查( , 一) 四肢 肌 力 和 肌 张 力 正 常 , 发 现 肌 肉假 性 肥 大 , 未 感 觉 正 常 , 理 反 射 存 在 , 理 反 射 未 引 出 . 助 检 查 , 酶 生 病 辅 肌
学 检 查 ; T2 / H H 8 / C 9 L, D 3 L. GO 9 U L, BD 3 2 U L, K 5 4 U/ L H 3 2U/
组型正常。
此家 系有 3代 共 计 l 人 , 中假 肥 大 型 肌 营 养 不 良症 患 7 其
者 2人 , 部是 男 性 患 者 , 系 调查 情 况详 见 图 1 全 家 。
J I I
2 讨 论
进 行 性 肌 营养 不 良症 的 病 因 和 发 病 机 制 复 杂 多 变 , 合 结 该 家 系 的 特 点 , 归 纳 如 下 : 均 在 男 性 身 上 发 病 , 始 发 病 可 ① 初
才 能 使 患 者 尽 可 能 地 从事 日常 活 动 。
3 参 考 文献
[]重 庆 医科 大 学 学报 ,07 3 (2 :38 J. 20 ,2 1)14 .
[ 稿 日期 :02 3 2 收 2 1 —0 —0 编校 : 林 ] 朱
[ ] 钟 敏 . 肥 大 型 肌 营 养 不 良症 基 因 缺 失 连 接 片 段 的 1 假
克 隆 和 应 用 [ ] 第一 军 医 大学 ,06 52 D. 200 1 .
。
[] 吴 志 英 , 2 陈雪 娇 . 营养 不 良分 子 遗 传 学 研 究 现 状 和对 肌
策 []中 华 神 经科 杂志 ,00 4 () 33 J. 2 1 ,3 5 :1 . [] 曹 俊 娟 , 3 柳 青 .uhne肌 营 养 不 良 症 的 研 究 进 展 D een
率 很 大 。 同 时 患 儿 家人 发 现 , 儿 小腿 变 粗 , 软 弱 无 力 。 连 患 但
穿 衣 、 臂 等 这 种 简 单 的 动 作 都 无 法 完 成 , 儿 抵 抗 力 和 机 体 抬 患 免疫力差 , 会伴随有咳嗽 、 常 咯痰 、 热 和 呼 吸 道 感 染 发 生 , 发 严
于女 性 , 好 发 于大 涎 腺 , 腺 约 占 8 % , 次 为 颌 下 腺 , 涎 且 腮 0 其 小 腺很少见。一般多为 单侧发病 ; I 表 现 为 缓 慢 增 长 的无 痛 临床 性肿物 , 近期 有 快 速 生 长 的 病 史 , 腺 L C应 注 意 与 鼻 咽 癌 、 涎 E
重 时 会有 明显 的气 促 和 呼 吸 困难 发 生 。 以 往 有 多 次 因 呼 吸 道
D D是 x一染 色 体 隐 性 遗 传 的 等 位 基 因病 , 致 病 基 因 M 其
主要 是 基 因 突 变 导 致 抗 肌 萎 缩 蛋 白 ( y r h ) 结 构 和 功 能 d so i 的 tp n
Ⅲ4为先 证 者 , ,O , 同龄 儿 童 比 较 , 小 肢 体 活 动 男 1岁 和 从
能力 弱 , 有 办 法 像 正 常 儿 童 那 样 正 常 行 走 、 动 , 至 4岁 没 活 长 多 才 能 够 慢 慢 独 立 行 走 , 速 度 仍 较 同 龄 人 慢 , 跌 倒 的 发 生 但 且
颌 下 腺 淋 巴上皮 癌 1例 报 告
齐玉英 , 权 国, 潘 卢 琴 ( 州 首 钢 水 钢 总 医 院 口腔 科 , 州 贵 贵 六 盘水 53 8 50 ) 2
[ 键 词 ] 颌下 腺 淋 巴上 皮 癌 ; 告 关 报
涎腺 淋 巴 上 皮 癌 ( m h phl a cl l,E ) 是 原 1 poeieal r i n L C , y t i e I 8 o 发 于 涎 腺 少 见 的 恶 性肿 瘤 之 一 , 占涎 腺 肿 瘤 的 O3 一04 。 .% .%