多环芳烃的测定----液相色谱法
HJ784-2016土壤和沉积物多环芳烃的测定高效液相色谱法方法验证报告

检测分析方法验证报告(验)字〔2022〕第号方法名称:土壤和沉积物多环芳烃的测定液相色谱法HJ 784-2016项目主编单位:验证单位:项目负责人及职称:(检测员)通讯地址:联系方式:报告日期:2022年03月17日土壤和沉积物多环芳烃的测定高效液相色谱法一、适用范围标准HJ 784-2016测定土壤和沉积物中十六种多环芳烃的液液萃取高效液相色谱法。
适用于土壤和沉积物中十六种多环芳烃的测定。
十六种多环芳烃(PAHs)包括:萘、苊、二氢苊、芴、菲、蒽、荧蒽、芘、苯并[a]蒽、屈、苯并[b]荧蒽、苯并[k]荧蒽、苯并[a]芘、茚并[1,2,3-cd]芘、二苯并[a,h]蒽、苯并[ghi]苝。
当取样量为10.0g,定容体积为1.0ml时,用紫外检测器测定16种多环芳烃的方法检出限为3μg/kg~5μg/kg,测定下限为12μg/kg~20μg/kg。
二、方法原理土壤和沉积物样品中的多环芳烃用合适的萃取方法(索氏提取、加压流体萃取等)提取,根据样品基体干扰情况采取合适的净化方法(硅胶层析柱、硅胶或硅酸镁固相萃取柱等)对萃取液进行净化、浓缩、定容,用配备紫外/荧光检测器的高效液相色谱仪分离检测,以保留时间定性,外标法定量。
三、试剂和材料3.1 乙腈(CH3CN):HPLC级。
3.2 正己烷(C6H14):HPLC级。
3.3 二氯甲烷(CH2Cl2):HPLC级。
3.4 丙酮(CH3COCH3):HPLC级。
3.5 丙酮-正己烷混合溶液:1+1。
用丙酮和正己烷按1:1的体积比混合。
3.6 二氯甲烷-正己烷混合溶液:2+3。
用二氯甲烷和正己烷按2:3的体积比混合。
3.7 二氯甲烷-正己烷混合溶液:1+1。
用二氯甲烷和正己烷按1:1的体积比混合。
3.8 干燥剂:无水硫酸钠(Na2SO4)或粒状硅藻土置于马弗炉中400℃烘4h,冷却后置于磨口玻璃瓶中密封保存。
3.9 硅胶:粒径75μm~150μm(200目~100目)。
多环芳烃的检测方法

多环芳烃的检测方法
1. 高效液相色谱法呀,这就像是一个超级侦探,能把多环芳烃从复杂的混合物中精准地揪出来!比如说在检测土壤中的多环芳烃时,它就能发挥大作用呢,难道不是很厉害吗?
2. 气相色谱-质谱联用法,哇哦,这简直就是检测多环芳烃的黄金搭档!就好像福尔摩斯和华生一样默契十足,能够准确地识别出多环芳烃的身份呢,你说神不神奇?
3. 荧光光谱法呢,就像一束神奇的光,能让多环芳烃无所遁形!在检测一些液体样品中的多环芳烃,效果那可是杠杠的,这多牛啊!
4. 免疫分析法,嘿,这可像个精准的小战士,专门对付多环芳烃!就拿检测食品中的多环芳烃来说,它可从来没让人失望过呀,是不是很赞?
5. 薄层色谱法,这看似简单却暗藏玄机,就如同一个低调的高手默默地工作着!想想看在一些快速检测的时候,它的作用可不小呢,难道不是吗?
6. 电化学分析法,哇,像是一个敏锐的传感器,能快速感知多环芳烃的存在!在一些特定环境的检测中,它可是立下了汗马功劳,真厉害呀!
7. 红外光谱法,像一双锐利的眼睛,能看穿多环芳烃的伪装!用于某些特定物质中的多环芳烃检测,那效果真是没得说,厉害吧!
8. 毛细管电泳法,好一个灵活的小能手,对付多环芳烃有一手!许多实验中它都表现出色,真让人佩服呢!
我觉得这些检测方法都各有千秋,在不同的场合和需求下都能发挥重要作用,我们真应该好好利用和研究它们,让多环芳烃无处遁形!。
水中的多环芳烃的测定

水中的多环芳烃的测定
多环芳烃是一类有机化合物,由若干个苯环组成,具有较高的毒性
和致癌性。
它们广泛存在于石油、煤炭等化石燃料中,也会通过工业
废水、农药、汽车尾气等途径进入水体中,对水环境造成严重污染。
因此,对水中多环芳烃的测定显得尤为重要。
一、测定方法
目前,常用的测定方法主要有气相色谱法、液相色谱法、质谱法等。
其中,气相色谱法是最常用的方法之一。
该方法利用气相色谱仪对样
品中的多环芳烃进行分离和检测,具有分离效果好、检测灵敏度高等
优点。
液相色谱法则是利用液相色谱仪对样品进行分离和检测,适用
于水中多环芳烃浓度较低的情况。
质谱法则是将气相色谱或液相色谱
与质谱联用,可以对多环芳烃进行定性和定量分析。
二、样品处理
在进行多环芳烃的测定前,需要对水样进行处理。
首先,需要将水样
进行预处理,去除其中的悬浮物和杂质。
其次,需要将水样进行萃取,将其中的多环芳烃提取出来。
萃取方法有多种,如固相萃取、液液萃
取等。
最后,需要对提取出来的多环芳烃进行纯化和浓缩,以提高检
测的灵敏度和准确性。
三、测定结果
通过对水样中多环芳烃的测定,可以得到其浓度和种类等信息。
根据测定结果,可以评估水体的污染程度,制定相应的治理措施。
同时,也可以为环境保护和水资源管理提供科学依据。
四、结论
水中多环芳烃的测定是一项重要的环境监测工作。
通过选择合适的测定方法和样品处理方法,可以得到准确可靠的测定结果。
这对于保护水环境、维护人类健康具有重要意义。
植物油中15种多环芳烃的高效液相色谱测定方法

植物油中15种多环芳烃的高效液相色谱测定方法目的建立植物油中15种多环芳烃的中性氧化铝小柱固相萃取—高效液相色谱检测方法。
方法植物油经正己烷溶解后过中性氧化铝固相萃取小柱净化,Waters PAH 4.6×250mm色谱柱进行分离,乙腈-水梯度洗脱,流速1.5ml/min,进样量10ul,柱温30,荧光检测器检测,外标法定量。
结果15种多环芳烃混合标准溶液在浓度为0.01-0.50ug/mL的范围内,在荧光检测器下呈良好的线性关系,该方法的检出限为0.1-1.5ug/kg之间,样品的加标回收率在62.14%-120.35%之间,RSD在0.91%-4.32%之间。
结论本实验所用方法具有样品前处理简单,检测方法高效快速,灵敏度高,准确性好等优点,能够满足植物油中多种多环芳烃含量的检测。
标签:多环芳烃;植物油;高效液相色谱法;食品安全Abstract:Objective To establish a method for simultaneous determination of 15 PAHs in plant oil by SPE-HPLC.Methods The edible vegetable oil was dissolved in n-hexaneand,cleaned up with neutral alumina SPE cartridges. The 15 PAHs was carried out by waters-PAHs column (4. 6 mm ×250 mm)with a gradient elution using acetonitrile-water as mobile phase at a flow rate of 1. 5 ml /min,the column temperature was 30 ℃,and the injection volume was 10 μl. Detection was carried out by a fluorescence detector with external standard.Results The 15 PAHs solution was a good linear relationship at a concentration within a range of 0.01-0.50ug / mL,The LOD was in the range of 0. 1 ~1.5 μg /kg with average recovery ranging from 62.14% to 120.35%. The RSD was in the range of 0. 91% ~4.32 0%.Conclusions The method had the advantages of simple pretreatment,high sensitivity and accuracy,which could be applied to the determination of the 15 PAHs in plant oil.Keywords:PAHs ;Edible vegetable oil ;HPLC;Food safety多环芳烃是一组由两个或两个以上苯环和稠环链接在一起的芳香族化合物及其衍生物,来源于工业生产、有机物热解或不完全燃烧等,为持久性有机污染物[1]。
液相色谱法测定水中16种多环芳烃的方法优化
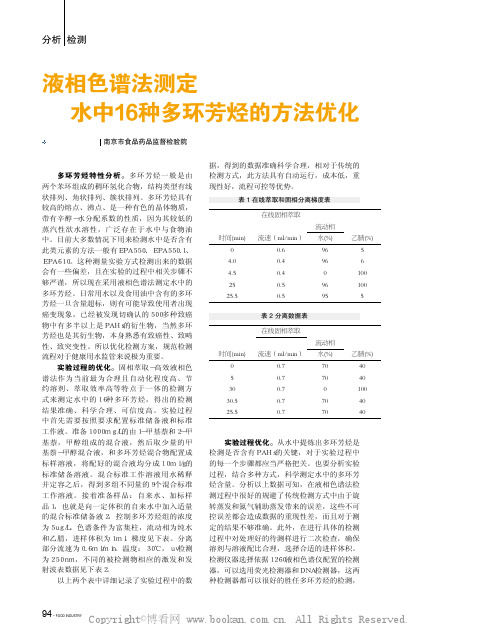
分析 检测液相色谱法测定水中16种多环芳烃的方法优化 应月 何苏 顾艳 南京市食品药品监督检验院实验测定过程及多环芳烃特性分析多环芳烃特性分析。
多环芳烃一般是由两个苯环组成的稠环氢化合物,结构类型有线状排列、角状排列、簇状排列。
多环芳烃具有较高的熔点、沸点、是一种有色的晶体物质,带有辛醇-水分配系数的性质,因为其较低的蒸汽性欲水溶性,广泛存在于水中与食物油中。
目前大多数情况下用来检测水中是否含有此类元素的方法一般有EPA550、EPA550.1、EPA610。
这种测量实验方式检测出来的数据会有一些偏差,且在实验的过程中相关步骤不够严谨,所以现在采用液相色谱法测定水中的多环芳烃。
日常用水以及食用油中含有的多环芳烃一旦含量超标,则有可能导致使用者出现癌变现象,已经被发现切确认的500多种致癌物中有多半以上是PAHs的衍生物,当然多环芳烃也是其衍生物,本身熟悉有致癌性、致畸性、致突变性。
所以优化检测方案,规范检测流程对于健康用水监管来说极为重要。
实验过程的优化。
固相萃取-高效液相色谱法作为当前最为合理且自动化程度高、节约溶剂、萃取效率高等特点于一体的检测方式来测定水中的16种多环芳烃,得出的检测结果准确、科学合理、可信度高。
实验过程中首先需要按照要求配置标准储备液和标准工作液。
准备1000mg/L的由1-甲基萘和2-甲基萘,甲醇组成的混合液,然后取少量的甲基萘-甲醇混合液,和多环芳烃混合物配置成标样溶液,将配好的混合液均分成10ml/g的标准储备溶液。
混合标准工作溶液用水稀释并定容之后,得到多组不同量的9个混合标准工作溶液。
接着准备样品:自来水、加标样品1,也就是向一定体积的自来水中加入适量的混合标准储备液2,控制多环芳烃组的浓度为5ug/L。
色谱条件为富集柱,流动相为纯水和乙腈,进样体积为1ml。
梯度见下表。
分离部分流速为0.6ml/min。
温度:30℃,uv检测为250nm,不同的被检测物相应的激发和发射波表数据见下表2。
环境空气和颗粒物中多环芳烃的测定高效液相色谱法

环境空气和颗粒物中多环芳烃的测定高效液相色谱法立题依据1)多环芳烃的理化性质多环芳烃(polycyclic aromatic hydrocarbons,PAHs)是一种含有两个或两个以上苯环或环戊二烯稠合而成的化合物。
包括稠环型和非稠环型两类,芳香稠环型是指分子中相邻的苯环至少有两个共用的碳原子的碳氢化合物,如萘、蒽、菲、芘等;芳香非稠环型是指分子中相邻的苯环之间只有一个碳原子相连的化合物,如联苯、三联苯等。
纯的PAHs通常是白色或浅黄绿色的固体,五环以上的PAHs大都是无色或淡黄色的结晶,个别具有深色,熔点及沸点较高,所以蒸气压低。
多环芳烃大多不溶于水,而辛醇-水分配系数比较高,易溶于苯类芳香性溶剂中。
多环芳烃大多含有π键,具有大的共轭体系,共轭体系中的电子具有较强的流动性,使得整个分子体系比较稳定,当多环芳烃发生化学反应时,趋向保留分子中共轭体系。
由于分子中存在高能反应键轨道π*和低能成键轨道π,当分子吸收了可见光或紫外光以后,价电子从成键轨道跃迁至返键轨道,当电子从激发态返回基态时,以荧光形式释放能量,形成特征吸收光谱和荧光光谱。
因此,多环芳烃具有一定荧光。
2)多环芳烃的主要来源绝大多数的多环芳烃在环境中不是单独存在,它们往往是两个或更多的多环芳烃的混合物。
多环芳烃大多是石油、煤等化石燃料以及木材、天然气、汽油、重油、有机高分子化合物、纸张、作物秸秆、烟草等含碳氢化合物的物质经不完全燃烧或在还原性气氛中经热分解而生成的,大都随烟尘、废气排放到空气,然后随空气沉降和迁移转化,进一步污染水体、土壤。
环境中多环芳烃的天然来源主要是陆地和水生生物的合成(沉积物成岩过程、生物转化过程、焦油矿坑内气体)、森林和草原火灾、火山爆发等过程中产生的,构成了多环芳烃的天然本地值。
环境中多环芳烃的主要来源是人为源。
人为源包括化学工业污染源、交通运输污染源、生活污染源和其他人为源。
木炭,原油,木馏油,焦油(天然),药物,染料,塑料,橡胶,农药(人为),润滑油,脱膜剂,电容电解液,矿物油,柏油(人为),杀虫剂、杀菌剂、蚊香、吸烟、汽油阻凝剂(人为)等都存在多环芳烃。
水质 多环芳烃的测定 液液萃取和固相萃取高效液相色谱法HJ478-2009 方法证实记录

水质多环芳烃的测定液液萃取和固相萃取高效液相色谱法HJ478-2009 方法证实记录一、方法原理用二氯甲烷萃取水中多环芳烃(PAHs),萃取液经弗罗里硅土柱净化,用二氯甲烷和正己烷的混合溶剂洗脱,洗脱液经浓缩后,用具有紫外检测器的高效液相色谱仪分离检测。
二、试剂2.1乙腈:SIGMA-ALDRICH,液相色谱纯2.2 甲醇:Fisher,液相色谱纯2.3 二氯甲烷:天津致远,色谱纯2.4 正己烷:SIMARK,液相色谱纯2.5 无水硫酸钠:天津光复,优级纯,400℃烘烤2h2.6 氯化钠:天津北联,优级纯,400℃烘烤2h2.7 多环芳烃标准溶液:SITAER,甲醇中16种多环芳烃混标(编号:301702F55)2.8 弗罗里硅土柱:100mg/6mL三、主要仪器设备3.1 液相色谱仪:Thermo,Μ-3000,配有紫外检测器3.2色谱柱:Acclaim TM 120 C18 5μm 120Å(4.6X250mm)3.3 分液漏斗:2000mL,活塞不加凡士林3.4 层析柱3.5旋转蒸发仪四、方法步骤4.1 样品预处理4.1.1 萃取摇匀水样,量取1000mL水样,倒入2000mL分液漏斗中,加入30g氯化钠,再加入50mL二氯甲烷,振摇5min,静置分层,收集有机相,放入250mL接收瓶中,重复萃取两遍,合并有机相,加入无水硫酸钠至有流动的无水硫酸钠存在。
放置30min,脱水干燥。
4.1.2 浓缩用旋转蒸发仪浓缩至1mL,加入正己烷至5mL,重复浓缩3次,最后浓缩至1mL,待净化。
4.1.3 净化用1g弗罗里硅土柱净化:先用4mL淋洗液(二氯甲烷:正己烷=1:1)冲洗净化柱,再用10mL正己烷平衡净化柱。
将浓缩后的样品加到柱上,再用3mL正己烷分3次洗涤装样品的容器,一并加到柱上,弃去流出的溶剂。
用10mL淋洗液洗涤净化柱,收集洗脱液于浓缩瓶中,浓缩至0.5~1.0mL后加入3mL乙腈,浓缩至0.5mL,最后用乙腈定容至1.0mL。
多环芳烃的测定----液相色谱法

多环芳烃的测定----液相色谱法1范围本法规定了用液相色谱分析法测定水中的萘(NPH)、荧蒽(FLU)、苯并(b)荧蒽(BbF)、苯并(k)荧蒽(BkF)、苯并(a)芘(BaP)、苯并(ghi)謋(BPer)和茚并(1,2,3,-cd)芘(IP)。
本法适用于供水和原水中多环芳烃(PAH S)的测定。
取水样500ml,将固相萃取洗脱浓缩到0.5ml,进样10μL,最低检测质量浓度(单位ng/L)为:NPH:35.5,FLU:1.2,BbF:1.7,BkF:0.05,BaP:1.0, Bper:1.3,IP:5.5。
2 原理硅胶基底的共价特性可使许多化学官能团(C8或C18)对其表面进行化学修饰,使水中半挥发、不挥发性有机污染物得以保留。
本法采用以粗颗粒(40μm左右)硅胶为基底的C18键合相作为固相吸附载体,对水中的PAH S进行吸附保留;用二氯甲烷等低极性有机溶剂洗脱PAH S后,用带紫外检测器的高效液相色谱仪进行定性和定量。
3 试剂3.1 流动相:甲醇和水3.1.1 甲醇:色谱纯,用前通过滤膜过滤和脱气。
3.1.2 水:用0.2μm滤膜过滤。
3.2 配制标准样品和水样预处理的试剂3.2.1 二氯甲烷:色谱纯。
3.2.2 四氢呋喃:色谱纯。
3.2.3 异丙醇:色谱纯。
3.2.4 硫代硫酸钠。
3.3 标准溶液:标准储备液。
4 仪器4.1 玻璃器皿所用玻璃器皿均需经铬酸洗液浸泡,洗净后自然晾干。
4.1.1 采样瓶:带磨口玻璃塞的棕色玻璃细口瓶。
4.1.2 尖底浓缩管:最小分度为0.1ml,容积必须进行标定,带磨口玻璃塞。
4.1.3 25μL微量注射器(液相色谱仪手下工进样器)。
4.1.4 量筒:50mL、100mL、和1000mL。
4.2 样品前处理装置4.2.1 固相萃取抽滤装置(负压)或恒流蠕动泵(正压)。
4.2.2 真空泵(30 L/min)。
4.2.3 SPE固相萃取柱:填料为40μm的C18键合相(500mg)吸附剂。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
多环芳烃的测定----液相色谱法1范围本法规定了用液相色谱分析法测定水中的萘(NPH)、荧蒽(FLU)、苯并(b)荧蒽(BbF)、苯并(k)荧蒽(BkF)、苯并(a)芘(BaP)、苯并(ghi)謋(BPer)和茚并(1,2,3,-cd)芘(IP)。
本法适用于供水和原水中多环芳烃(PAH S)的测定。
取水样500ml,将固相萃取洗脱浓缩到0.5ml,进样10μL,最低检测质量浓度(单位ng/L)为:NPH:35.5,FLU:1.2,BbF:1.7,BkF:0.05,BaP:1.0, Bper:1.3,IP:5.5。
2 原理硅胶基底的共价特性可使许多化学官能团(C8或C18)对其表面进行化学修饰,使水中半挥发、不挥发性有机污染物得以保留。
本法采用以粗颗粒(40μm左右)硅胶为基底的C18键合相作为固相吸附载体,对水中的PAH S进行吸附保留;用二氯甲烷等低极性有机溶剂洗脱PAH S后,用带紫外检测器的高效液相色谱仪进行定性和定量。
3 试剂3.1 流动相:甲醇和水3.1.1 甲醇:色谱纯,用前通过滤膜过滤和脱气。
3.1.2 水:用0.2μm滤膜过滤。
3.2 配制标准样品和水样预处理的试剂3.2.1 二氯甲烷:色谱纯。
3.2.2 四氢呋喃:色谱纯。
3.2.3 异丙醇:色谱纯。
3.2.4 硫代硫酸钠。
3.3 标准溶液:标准储备液。
4 仪器4.1 玻璃器皿所用玻璃器皿均需经铬酸洗液浸泡,洗净后自然晾干。
4.1.1 采样瓶:带磨口玻璃塞的棕色玻璃细口瓶。
4.1.2 尖底浓缩管:最小分度为0.1ml,容积必须进行标定,带磨口玻璃塞。
4.1.3 25μL微量注射器(液相色谱仪手下工进样器)。
4.1.4 量筒:50mL、100mL、和1000mL。
4.2 样品前处理装置4.2.1 固相萃取抽滤装置(负压)或恒流蠕动泵(正压)。
4.2.2 真空泵(30 L/min)。
4.2.3 SPE固相萃取柱:填料为40μm的C18键合相(500mg)吸附剂。
4.3 高压液相色谱系统4.3.1 HPLC恒流泵:流速精度为0.01 mL/min。
4.3.2 色谱柱:反相98(ODS)分析柱,推荐柱长为150mm,内径为4.6mm,粒度为5μm 并配备40mm相同填料的预分离柱。
4.3.3 荧光检测器:具有激发/发射光谱扫描功能,同时具有波长程序设置功能。
4.3.4 紫外检测器:可单独使用,也可与荧光检测器串联使用。
4.3.5 数据处理系统:色谱工作站或积分仪。
5 样品5.1 样品瓶的准备用干净的棕色玻璃瓶作为采样瓶。
5.2 水样采集样品必须采集在棕色玻璃瓶中,若水中有残余氯,需在每升水中加入25mg的硫代硫酸钠除氯;若不能立即进行样品处理,建议在采样时每升水样加入200mL的异丙醇作为样品稳定剂(同时也作为基体改性剂)。
5.3 水样保存水样应置于暗处,4℃冰箱中保存,应在24h内尽快进行样品预处理。
将吸附后的固相萃取柱直接贮存在冰箱中,在20天内将PAHs从固体萃取柱上洗脱下来,进行样品分析。
5.4 样品预处理5.4.1 水样准备:量取500~2000mL水样(根据配备的液相色谱仪灵敏度和水源污染状况,可选择合适的水样体积)。
每1L水样,加入200ml的异丙醇(也可在采样时加入),混合均匀。
5.4.2 SPE柱活化及条件化:先用2ml二氯甲烷注入柱子,让其缓缓流过,并抽空气5min,以去除填料中可能存在的干扰物,再加入2ml的甲醇活化柱子,最后用去离子水(加入和水样相同含量的改性剂)移去活化溶剂(条件化),但注意在对SPE柱进行活化和条件化时,不可将柱床抽干,以防止填料层产生裂隙,使回收入率降低。
5.4.3 水样富集:以4~5ml/min左右的流速,使水样全部通过SPE柱后,加入5ml纯水,让其缓缓通过,以去除水溶性干扰物质,通入净化空气30min,使已吸附的SPE柱彻底干燥。
5.4.4 洗脱与浓缩:将2ml二氯甲烷或四氢呋喃分两次加入柱管,分别洗下被测组分,合并后的洗脱液用氮气浓缩至0.1ml以下,再定容至0.1~0.5ml,待进样分析。
6 测定步骤6.1 色谱条件的选择(根据仪器型号、配置状况,选择合适的色谱条件)6.1.1 流动相:甲醇和水。
根据色谱柱的柱效和分离度,选择合适的流动相配比。
如果用甲醇/水体系能满足样品的分离要求,就不必再用其他流动相体系,如果用纯甲醇即可达到分离效果,就不要用水作为流动相成分。
6.1.2 洗脱模式及洗脱流速:采用等度洗脱,流动相为甲醇:水=80:20,洗脱速率为1.0ml/min。
如果分离效果不理想,适当增加流动相中水的比例,或考虑梯度洗脱。
6.1.3 柱箱温度控制:使用色谱柱温箱,柱箱温度为35℃。
6.1.4 检测器波长设置6.1.4.1 荧光检测器:本方法首先进行激发和发射光谱的扫描,根据所获光谱图,找出各化合物的特征激发和发射波长,见表1。
表1 多环芳烃最佳的荧光激发和发射波长PSHs 激发波长λex,mm 发射波长λex,mmFLU 226 449BbF 302 452BkF 302 431BaP 297 405Bper 302 420IP 305 500 为获得较高方法的灵敏度,根据最优化柱系统条件下获得的各PAHs的保留时间,进行波长程序设置,以便使其能在最优化的激发和发射波长下进行检测。
6.1.4.2 紫外检测器:检测波长为254nm(如果当地水源中背景干扰严重,影响了检测,可选用其他紫外波长)。
6.1.5 数据记录和处理系统:各型色谱工作站均可使用,色谱工作站兼有积分仪功能。
同时还具有双通道数据采集、谱图再处理及精确的定性定量分析等功能。
若采用积分仪,要进行合理的参数设置,以便记录下理想的谱图。
6.2 标准曲线的绘制本方法使用外标法定量。
6.2.1 标准样品的制备根据液相色谱仪的线性工作范围,选择不同浓度的标准工作溶液(至少5个点),所用标准工作液由混合的PAHS标准液用甲醇稀释制得。
浓度系列NPHmg/LFLUμg/LBbFμg/LBkFμg/LBaPμg/LBPerμg/LIPμg/L1 2 3 4 5 2.04.06.08.010.020.040.060.080.0100.08.016.024.032.040.08.016.024.032.040.020.040.060.080.0100.032.064.096.0128.0160.020.040.060.080.0100.0相关系数0.999 0.999 0.999 0.997 0.998 0.996 0.999 6.2.2 标准液的使用每个工作日必须测定一种或几种浓度的标准溶液以检验标准曲线,若某一化合物的测定值与期望值的相对标准偏差大于10%,则必须重新绘制标准曲线。
6.2.3 标准曲线的表示以响应值对浓度作标准曲线,由回归方程计算测定结果。
若采用工作站,只需按已设定的样品序列依次进样,计算机自动存储标准曲线,自动计算每一次进样的测定结果。
若采用积分仪,可采用常规标准曲线绘制和样品测定结果的计算方式。
6.3 样品测定6.3.1 进样6.3.1.1 进样方式:以注射器人工进样或自动进样器进样。
6.3.1.2 进样量:5~25μL。
6.3.1.3 操作:用待测试样调湿微量注射器针头及针筒,并洗涤三次,缓缓反复多次尽可能排出针筒内气泡,迅速注射样品至HPLC柱头,进行HPLC分析,并用甲醇洗涤注射器,以备下次进样。
6.3.2 记录:由色谱工作站完成。
6.4 定性分析6.4.1 保留值定性法。
6.4.2 鉴定的辅助方法。
6.5 定量分析用外标法计算。
7 计算采用色谱工作站,计算出各组分的含量,单位μg/L。
样品浓度:ρi=ρ0V t/V S式中:ρi——试样中组分质量浓度,μg/L。
ρ0——固相萃取洗脱液质量浓度,μg/L。
V t——固相萃取洗脱液浓缩后定容体积,mL。
V S——水样体积,mL。
如果采用积分仪,样品浓度:ρi=A i B i V t/V S V i式中:ρi——试样中组分质量浓度,μg/L。
A i——试样中组分的进样量对峰高(或峰面积)比值,ng/mm(ng/mm2)。
B i——样品中组分峰高(或面积),mm(mm2)。
V t——萃取液浓缩后体积,μL。
V i——注射样品的体积,μL。
V S——水样体积,mL。
8 注意事项8.1 使用标准样品条件8.1.1 标准样品进样体积与试样体积相同,标准样品浓度应接近试样的浓度。
8.1.2 标准样品和试样尽可能同时分析,直接与单个标样比较以测定浓度。
8.2 安全8.2.1 所用有机溶剂甲醇有毒性,四氢呋喃、正己烷易燃,均为易挥发性试剂,操作时必须遵守有关规定,重蒸馏有机溶剂必须在通风柜中进行,严禁明火。
8.2.2 分析的PAH S为致癌物,因此要有保护措施。
8.2.3 用过的废液集中处理后排放。
戴安高效液相色谱性能指标:1、P680 HPG低压四元梯度泵:双柱塞串联方式,采用等动力学预压缩技术(Gynkotek专利),提供无脉冲流动相且无需阻尼器,对高效液相色谱系统的重现性和性能都有显著的提高;自动液漏检测。
(1)、具有溶剂自动补偿功能;延迟体积:<400ul;压力范围:0~7250psi(500bar);(2)、压力脉冲:<1%;流速精度:±0.1%,at±1ml/min;(3)、流量范围:0.001~10ml/min,0.001ml/min增量;2、UV170检测器:四波长检测技术,可升级到二极管阵列检测器。
(1)、灯源:氘灯;波长范围:200~595nm;(2)、噪声:±5×10-6AU(干电池噪声254 nm,1秒);(3)、光谱带宽:1.9~400nm,程序可调;波长精度:±0.75 nm;(4)、波长校正:内置氧化钬滤光片(符合ASTM标准)。
液相色谱操作规程一、电源检查各设备是否联接好二、仪器开机1、P680电源接通后,根据实验要求(国家标准方法)设定参数。
1)、流动相流速按Flow键后,按数字键输入,按Enter键确认。
2)、 混合比例按A/B/C/D键后,在压力显示窗口显示所选通道的混合比例。
按数字键输入,按Enter 键确认。
注意:A、B、C、D四个通道混合比例的代数和必须是1。
3)、 压力极限按Max/Min键后,按数字键输入,按Enter键确认。
如果P680在运行时高于高压极限或者低于低压极限,停泵的同时,Off指示灯亮,A、B、C、D、MIN、MAX指示灯闪亮;P680和CHROMELEON的屏幕中也将出现相应的错误提示。
2、启柱温箱,根据要求设定温度。
3、开启检测器,氘灯不会自动地开。
灯的开/关必须通过CHROMELEON TM执行,为了获得最佳的结果,灯必须在分析前30分钟开。