化妆品中α氯甲苯的检测方法
化妆品的卫生化学检测方法

化妆品的卫生化学检测方法
重金属检测是为了确保化妆品产品中的重金属元素含量符合卫生标准。
检测方法包括原子吸收光谱法(AAS)、电感耦合等离子体质谱法(ICP-MS)和电感耦合等离子体发射光谱法(ICP-OES)等。
这些方法以其高灵
敏度、高选择性和高准确性在化妆品行业得到广泛应用。
残留物检测主要是检测化妆品中残留的药物成分、有害物质、添加剂
和污染物等。
常见的检测方法包括气相色谱法(GC)、液相色谱法(HPLC)和紫外-可见分光光度法等。
这些方法通过提取样品中的有机物或无机物,然后以色谱或光谱技术进行分离和鉴定。
挥发性有机物检测主要是检测化妆品产品中的甲醛、甲苯等有害挥发
性有机物。
检测方法包括气相色谱-质谱联用法(GC-MS)、气相色谱法(GC)和红外光吸收法等。
这些方法通过灵敏的仪器分析技术,可以快速、准确地检测出化妆品中的有害挥发性有机物。
除了以上几个方面的卫生化学检测外,化妆品还需要进行其他物理性
和理化性的测试,例如颗粒度分析、粘度测定、pH值测定、保质期测试等。
这些测试方法可以帮助生产企业确保化妆品产品的质量和安全性。
总之,化妆品的卫生化学检测是确保化妆品产品质量和安全的重要手段。
各种化学检测方法的应用可以全面、准确地评估化妆品产品的卫生性能,为消费者提供安全、可靠的化妆品产品。
化妆品中挥发性有机溶剂检测方法
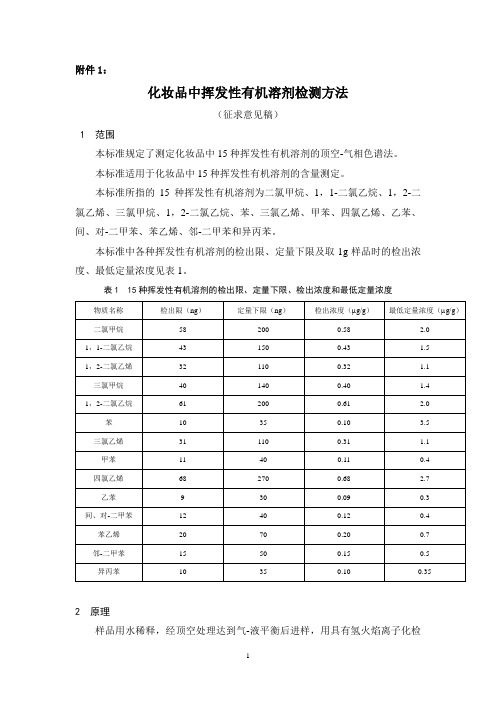
附件1:化妆品中挥发性有机溶剂检测方法(征求意见稿)1 范围本标准规定了测定化妆品中15种挥发性有机溶剂的顶空-气相色谱法。
本标准适用于化妆品中15种挥发性有机溶剂的含量测定。
本标准所指的15种挥发性有机溶剂为二氯甲烷、1,1-二氯乙烷、1,2-二氯乙烯、三氯甲烷、1,2-二氯乙烷、苯、三氯乙烯、甲苯、四氯乙烯、乙苯、间、对-二甲苯、苯乙烯、邻-二甲苯和异丙苯。
本标准中各种挥发性有机溶剂的检出限、定量下限及取1g样品时的检出浓度、最低定量浓度见表1。
表1 15种挥发性有机溶剂的检出限、定量下限、检出浓度和最低定量浓度2 原理样品用水稀释,经顶空处理达到气-液平衡后进样,用具有氢火焰离子化检测器的气相色谱仪进行分析,以保留时间定性,峰面积外标法定量。
3试剂3.1甲醇,色谱纯。
3.2氯化钠,分析纯:550℃烘2h~3h。
3.315种挥发性有机溶剂标准溶液:分别称取15种挥发性有机溶剂标准品各10mg(精确至0.1mg),分别置于已加少量甲醇的10mL容量瓶中,待溶解完全后用甲醇定容。
配成如表2所示浓度的标准储备溶液单标,再取各标准储备溶液单标适量,用水稀释配成混合标准使用溶液和标准系列。
表2 15种挥发性有机溶剂的标准储备溶液浓度及标准系列浓度4 仪器4.1气相色谱仪,具氢火焰离子化检测器,分流/不分流进样口,配色谱工作站。
4.2自动顶空装置,或超级恒温水浴锅(控温精度 0.5℃)和气密针。
4.3顶空瓶(20mL)。
5分析步骤5.1 样品预处理称取样品约1.0g(精确至1mg)于100mL具塞刻度管中,加入水至刻度,混匀,此溶液作为待测样液备用。
5.2 测定5.2.1 色谱参考条件色谱柱:DB-1,30m×0.32mm I.D.,0.25 m。
氮气流速:45.0mL/min;氢气流速:40.0mL/min;空气流速:450mL/min。
分流比:1:10。
柱流量:1.0mL/min。
检测器:氢火焰离子化检测器。
化妆品32种禁限用染成分的检测方法和编制说明

化妆品32种禁限用染成分的检测方法和编制说明化妆品 32 种禁限用染料成分的检测方法和编制说明附件 2: 化妆品中 32种禁限用染料成分的检测方法 1 范围本方法规定了测定染发类化妆品中 32 种禁限用染料成分(见附录 A)的高效液相色谱法。
本方法适用于染发类化妆品中 32 种禁限用染料成分的含量测定。
本方法无法区分所涉及染料成分的游离态、硫酸盐和盐酸盐,化妆品中各种形态同时存在时,应换算成一种形态,以总量表示。
2 原理用无水乙醇水11 的混合溶液提取染发类化妆品中的 32 中禁限用染料成分,用高效液相色谱仪进行分析,以保留时间和紫外吸收光谱定性,峰面积定量。
本方法中 32 种禁限用染料成分的检出限、定量下限及取 0.5g 样品时的检出浓度及最低定量浓度见表 1。
表 1 32 种禁限用染料成分的检出限、检出浓度、定量下限、最低定量浓度 - 1 - - 2 - 3 试剂除另有规定外,所用试剂均为分析纯,水为超纯水。
3.1 无水乙醇。
3.2 甲醇,色谱纯。
3.3 乙腈,色谱纯。
3.4 亚硫酸氢钠。
3.5 磷酸溶液(19):吸取磷酸(ρ201.69g/mL)10mL,加水 90mL。
3.6 磷酸盐混合溶液:称取十二水合磷酸氢二钠 1.8g、磷酸二氢钾 2.8g 和庚烷磺酸钠(C7H15SO3Na)1.0g,用水稀释至 1L,混匀,配制成含庚烷磺酸钠(1g/L)的磷酸盐缓冲液,加入磷酸溶液(3.5),调节 pH 至 6.0 左右,0.45m 微孔滤膜过滤。
3.7 染料类化合物标准储备溶液(ρ(染料成分)10g/L):称取染料对照品约 100mg(精确至 0.1mg)于 10mL 容量瓶中,以 2g/L 亚硫酸氢钠水溶液无水乙醇(3.1)11的混合溶液定容至 10mL,配成约 10g/L 的单标溶液。
以下几种物质在上述溶剂中的溶解性较差,分别采取如下措施:甲苯-25-二胺硫酸盐和 2-氯-p-苯二胺硫酸盐 2 种物质直接用2g/L 亚硫酸氢钠水溶液溶解并定容;甲苯-34-二胺直接用无水乙醇(3.1)定容;2-硝基-p-苯二胺和 4-硝基-o-苯二胺需将称样量减至 25mg,再用无水乙醇(3.1)定容,配成约2.5g/L 的单标溶液。
化妆品中36种挥发性有机溶剂残留的测定

化妆品中36种挥发性有机溶剂残留的测定蒋凯;薛晓康;李晓宇【摘要】建立双柱定性-顶空气相色谱质谱联用仪法测定化妆品中36种挥发性有机溶剂残留的方法.使用极性柱VF-1301 ms和非极性柱DB-5 ms两根色谱柱,考察挥发性有机溶剂残留的保留时间以定性,再利用VF-1301 ms柱对存在的残留溶剂进行定量测定.所建立的方法在相应的浓度范围内浓度和峰面积的线性关系良好,线性相关系数为0.999 1~0.999 7,加标回收率在82.0%~117.6%之间,相对标准偏差为1.1%~2.9%,检出限为0.08~29.24 μg/g.该方法检测结果准确、可靠,适用于多种常用化妆品挥发性有机溶剂的同时测定.【期刊名称】《应用化工》【年(卷),期】2019(048)003【总页数】4页(P728-731)【关键词】双柱定性;挥发性有机溶剂;化妆品;顶空气相色谱-质谱法【作者】蒋凯;薛晓康;李晓宇【作者单位】上海化学品公共安全工程技术研究中心上海化工研究院有限公司,上海200062;上海化学品公共安全工程技术研究中心上海化工研究院有限公司,上海200062;上海化学品公共安全工程技术研究中心上海化工研究院有限公司,上海200062【正文语种】中文【中图分类】TQ658;O658随着人们生活水平的提高,化妆品在人类的日常生活中也逐渐占据了不可替代的位置。
而化妆品中普遍存在挥发性有机溶剂残留,这些有机溶剂常用于溶解和分散杀菌防腐剂、香精、表面活性剂油脂及颜料等组分[1-3]。
如长期使用和接触这些有机溶剂,会对人体产生相应的毒害作用,如对皮肤、眼睛和呼吸道造成刺激作用,麻痹和损伤神经,损伤皮脂层等等[4]。
因此,同时对化妆品中多种挥发性有机溶剂残留的测定方法是十分必要的。
然而化妆品成分大多十分复杂,含有多种香精香料,在分析时常常造成干扰。
本文采用双柱定性-顶空气相色谱质谱法[5-15],对36种常见的挥发性有机溶剂进行了测定。
化妆品中少量氯化物的测定

2.为什么要做空白试验?滴定过程中为何要用 力摇动? 一般试验都要设置参比试验,为的就是 控制一个变量,消除其他变量带来的对试验 结果的影响。
3.以K2CrO4作指示剂时,指示剂的浓度过大 或过小对测定有何影响? 指示剂浓度过大,则测量得到的CL的实 际值变小,例如假设17%浓度的指示剂中, 正常反应氯一摩尔,此时由于指示剂浓度过 大,实际反应的数量超过一摩尔,测得的值 纠偏小了,反之亦然。
④计算结果
Cl-(mg/L) =
(V2-V1)×C×35.5×1000 -----------------------V水
C—硝酸银标准溶液浓度(mol/L); V水—水样的体积(mL); V1——蒸馏水消耗硝酸银溶液的体积(ml); V2——水样消耗硝酸银标准溶液的体积(ml)。
1.滴定中试液的酸度宜控制在什么范围内?为 什么?怎样调节? 严格控制酸度,酸度大了会引起酸效应, 使络合不完全;酸度小了常温不能反应,准 确控制酸度的方法是一般加入缓冲溶液。调 节酸碱度应用弱酸或弱碱,避免发生其他反 应。
化妆品中少量 氯化物的测定
目浊法 沉淀滴定法
一.目浊法
1.原理:试样溶液中含有微量氯离子与硝酸银 生成白色的氯化银沉淀,其浊度与标准氯离 子产生的氯化银比较,进行目视比浊。
ห้องสมุดไป่ตู้
2.操作步骤 ①称取试样10g,精密至0.1g,加水溶解并定容 至100ml,摇匀。 ②吸取试样液10.00ml于一只50ml纳氏比色管中, 加水13ml,摇匀;再准确吸取氯化物标准溶液 10.00ml于另一只纳氏比色管中,加水13ml,摇 匀,同时向上述两管加硝酸溶液(10%)和硝酸 银标准溶液【C(AgNO3)=0.1mol/L】各1ml,立 即摇匀,于暗处放置5min后取出立即进行目视 比浊。
《化妆品中挥发性有机溶剂检测方法》

《化妆品中挥发性有机溶剂检测方法》化妆品是现代人日常生活中必不可少的用品之一,而其中也包含了许多挥发性有机溶剂。
这些溶剂可以在化妆品的生产、运输和使用过程中发挥重要作用,但如果使用不当或导入过多,就可能对人体健康产生一定的潜在危害。
因此,发展一种简便、快速、准确的挥发性有机溶剂检测方法具有重要意义。
目前,对于挥发性有机溶剂的检测,常常采用气相色谱法(Gas Chromatography,GC)和质谱法(Mass Spectrometry,MS)进行分析。
GC-MS方法具有分辨率高、灵敏度高、选择性好等优点,因此广泛应用于挥发性有机溶剂的快速分析。
该方法通过高分辨率气相色谱对溶剂进行分离,然后通过质谱进行定性和定量分析。
然而,GC-MS方法分析过程繁琐,需要复杂的样品预处理过程,且需要高昂的设备和操作技术。
因此,需要开发更加简捷、经济、环保的挥发性有机溶剂检测方法。
通过对文献进行综合分析,发现有一种基于红外光谱技术的挥发性有机溶剂检测方法具有潜在的应用前景。
红外光谱分析是一种非破坏性的分析方法,可快速获取样品的红外光谱图谱,并通过谱图中的特征峰进行溶剂组分的定性和定量分析。
此外,该方法的仪器简单,操作便捷,无需昂贵的设备和复杂的样品预处理过程,具有广泛应用的潜力。
基于红外光谱的挥发性有机溶剂检测方法主要有两种,分别是红外吸收光谱法(Infrared Absorption Spectroscopy)和红外传感器法(Infrared Sensor)。
前者通过红外光的能量吸收特性来实现溶剂的鉴定和定量分析,后者则通过红外传感器对红外光的反射或透射进行检测,从而实现溶剂的快速检测。
红外光谱的挥发性有机溶剂检测方法已在实际应用中取得较好的效果。
例如,在化妆品生产过程中,可以通过红外光谱分析仪对不同工序的溶剂进行实时监测,确保在生产过程中溶剂的控制达到要求。
此外,在化妆品质量控制方面,也可以利用红外光谱分析方法进行关键溶剂成分的定量测定,保证产品的质量稳定性。
化妆品中毒化物检测方法

化妆品中毒化物检测方法化妆品在现代社会中被广泛应用,给人们带来美丽的同时也带来了一定的安全隐患。
化妆品中含有毒性物质或过量使用可能对健康造成危害。
因此,针对化妆品中毒化物的检测方法变得非常重要。
在本文中,将介绍几种常用的化妆品中毒化物检测方法。
一、高效液相色谱法(HPLC)高效液相色谱法是一种常用于化妆品中毒化物检测的准确可靠的方法。
该方法利用高效液相色谱仪将化妆品中的化学物质分离并定量分析。
首先,将样品溶解在适当的溶剂中,并通过进样器进入色谱系统。
然后,通过不同组分在色谱柱上的保留时间和吸光度信号,可以确定化妆品中的毒性物质含量。
高效液相色谱法具有准确、灵敏、快速、复现性好的优点,因此被广泛应用于化妆品中毒化物的检测。
二、气相色谱质谱联用法(GC-MS)气相色谱质谱联用法是一种强大的分析工具,可以用于检测化妆品中的毒性物质。
该方法首先通过气相色谱仪将样品中的化学物质分离,然后将分离的化学物质与质谱联用进行进一步检测和鉴定。
气相色谱可以分离非极性或低极性化合物,而质谱可以对物质的分子结构进行定性和定量分析。
因此,气相色谱质谱联用法能够快速准确地确定化妆品中的毒性物质成分。
三、光谱方法(UV-Vis、FTIR)光谱方法是一种常见的化学分析方法,通过物质与光的相互作用,检测和分析样品中的成分。
在化妆品中毒化物的检测中,UV-Vis光谱和FTIR(傅里叶红外)光谱是常用的方法。
UV-Vis光谱法利用化妆品中毒化物与特定波长的光发生吸收作用,通过测量吸收光强度可以确定物质的浓度。
FTIR光谱则通过检测化妆品中毒化物吸收红外光的特定波长来鉴定化合物的结构。
这两种方法都具有操作简单、快速、可靠的优点,广泛应用于化妆品中毒化物的分析。
四、质量谱法(MS)质谱法是一种高灵敏度、高选择性的分析方法,常用于化学物质的鉴定和分析。
质谱仪通过对样品中的化学物质进行高能量撞击,将分子分解成离子,并通过离子的质量分析确定化合物的质量和结构。
化妆品成分检测方法

化妆品成分检测方法化妆品是人们日常生活中常见的美容产品,而了解其成分的安全性和质量是很重要的。
因此,建立一套可靠的化妆品成分检测方法是非常必要的。
本文将介绍一些常见的化妆品成分检测方法,包括化学分析法、生物学分析法、物理分析法等。
一、化学分析法化学分析法是通过化学反应、化学物性等方法来定性和定量分析化妆品成分的方法。
其中,常用的化学分析方法包括色谱法、质谱法、核磁共振法等。
这些方法可以对化妆品进行快速准确的成分检测和分析。
色谱法是目前最常用的化学分析方法之一。
它可以根据化妆品中成分的不同极性和挥发性,在色谱柱中进行分离和检测。
常见的色谱方法包括气相色谱法(GC)和液相色谱法(LC)。
通过色谱法可以准确分离化妆品中的各种有机成分,并确定其成分浓度。
质谱法是一种能够精确测量化妆品成分相对分子质量和结构的化学分析方法。
常见的质谱方法包括质谱联用技术(GC-MS、LC-MS)和飞行时间质谱法(TOF-MS)。
质谱法可以提供化妆品成分的详细信息,帮助确定其组成和来源。
核磁共振法是利用核磁共振现象来研究化学物质结构和性质的方法。
在化妆品成分分析中,常用的核磁共振技术包括核磁共振波谱法(NMR)和质子磁共振谱法(1H-NMR)。
核磁共振法可以帮助确定化妆品成分的分子结构和相对含量。
二、生物学分析法生物学分析法主要是通过生物活性试验和皮肤刺激试验等方法来检测化妆品成分的安全性和效果。
其中,常用的生物学分析方法包括细胞毒性试验、动物试验和人体试验。
细胞毒性试验是通过将化妆品成分暴露在细胞培养基上,观察细胞的生存和增值情况,进而评估成分对细胞的毒性。
这种试验可以帮助判断化妆品成分对细胞的损伤程度。
动物试验是将化妆品成分应用于动物身上,观察其对动物的毒性和刺激作用。
然而,由于动物试验存在伦理问题,现今已逐渐减少在化妆品成分检测中的应用。
人体试验是将化妆品成分应用在志愿者的皮肤上,观察其对皮肤的刺激性和过敏性。
这种试验可以更真实地评估化妆品成分对人体的影响。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
附件7
化妆品中α-氯甲苯的检测方法
1 适用范围
本方法规定了采用气相色谱法测定化妆品中禁用物质α-氯甲苯(CAS:100-44-7)的方法。
本方法适用于洗护发类、膏霜类、乳液类、化妆水类化妆品中α-氯甲苯的测定。
2 方法提要
样品在经过提取后,采用气相色谱仪分离,用氢火焰离子化检测器检测。
根据保留时间定性,峰面积定量,以标准曲线法计算含量。
必要时,采用气相色谱-质谱(GC-MS)确证。
本方法对α-氯甲苯的检出限为0.00054μg,定量下限为0.0018μg。
若取2.0g样品,本方法对α-氯甲苯的检出浓度为2.7μg/g,最低定量浓度为9μg/g。
3 试剂和材料
除另有规定外,所用试剂均为分析纯。
水为一级实验用水。
3.1 三氯甲烷(色谱纯)。
3.2 α-氯甲苯,纯度≥99%。
3.3 正己烷(色谱纯)。
3.4 饱和氯化钠溶液:称取40 g 氯化钠,置于250mL磨口锥形瓶中,加入100 mL水,超声15分钟,即得。
3.5 无水硫酸钠。
3.6 α-氯甲苯标准储备溶液:称取α-氯甲苯(3.2)0.1g,精确至0.0001g,置100mL容量瓶中,用三氯甲烷(3.1)溶解并稀释至刻度,摇匀,配成质量浓度为1g/L的标准储备溶液。
3.7 α-氯甲苯标准工作溶液:用三氯甲烷(3.1)将上述标准储备溶液(3.6)分别配成α-氯甲苯浓度为2.5μg/mL、12.5μg/mL、25μg/mL、50μg/mL、100μg/mL的标准工作溶液。
4 仪器
4.1 气相色谱仪:具氢火焰离子化检测器。
4.2 气相色谱-四极杆质谱联用仪(GC-MS)。
4.3 分析天平:感量0.0001g。
4.4 分析天平:感量0.01g。
4.5 离心机。
5 测定步骤
5.1 样品前处理
称取样品约2g,精确至0.0001g,置于100mL具塞锥形瓶中,加入10mL饱和氯化钠溶液(3.4),充分振摇,使样品分散后转移至25mL分液漏斗,加5mL三氯甲烷(3.1),振摇提取30s,静置分层,将三氯甲烷提取液放入10mL具塞比色管,水相加三氯甲烷(3.1)重复提取步骤一次,合并二次三氯甲烷提取液,补加三氯甲烷(3.1)至刻度,加入适量无水硫酸钠(3.5)脱水(必要时取提取液,5000r/min离心5min,取上清液),溶液经0.45μm滤膜过滤,取续滤液作为待测溶液,备用。
5.2 测定
5.2.1 气相色谱(GC-FID)参考条件
色谱柱:DB-1701P(30m×0.32mm×0.25μm)或相当极性的毛细管色谱柱;
柱温程序:90℃(10min),10℃/min 升至250℃(10min);
进样口温度:200℃;
检测口温度:250℃;
载气:N2,流速:1.5mL/min;
氢气流量:40mL/min;
空气流量:400mL/min;
尾吹氮气流量:25mL/min;
进样方式:分流进样,分流比5:1;
进样量:1μL。
5.2.2 测定方法
按“,取α-氯甲苯标准工作溶液(3.7)分别进样,进行气相色谱分析,以峰面积为纵坐标,标准溶液浓度为横坐标进行线性回归,建立标准工作曲线。
取“5.1”项下处理得到的待测溶液进样,根据测定成分的峰面积,代入标准工作曲线得出α-氯甲苯的质量浓度。
按“6 计算”,计算试样中α-氯甲苯的质量分数。
5.3 平行实验
按以上步骤操作,对同一样品独立进行测定获得的两次独立测试结果的绝对差值不得超过算术平均值的10%。
5.4 阳性结果确证
测定过程中若有阳性结果,可采用气相色谱-质谱法进一步确证。
在相同的实验条件下,如果样品中检出的色谱峰的保留时间与标准溶液中对应成分一致,所选择的监测离子的相对丰度比与相当浓度标准溶液的选择监测离子相对丰度比的偏差不超过表1规定范围,则可以判定样品中存在对应的待测成分。
试样溶液中α-氯甲苯含量较高时,可用三氯甲烷(3.1)稀释制成含α-氯甲苯为0.2μg/mL~12μg/mL的浓度范围进行气相色谱-质谱法测定。
表1 阳性结果确证时相对离子丰度比的最大允许偏差
色谱柱:VF-1701MS(30m×0.25mm×0.25μm)或相当极性的毛细管色谱柱;
柱温程序:90℃(10min),10℃/min 升至250℃(10min);
进样口温度:200℃;
载气:氦气1.0mL/min;
进样方式:分流进样,分流比5:1;
进样量:1μL。
电离方式:电子轰击源(EI),电离能量为70eV。
离子源温度:220℃。
传输线温度:220℃。
溶剂延迟时间:4分钟。
扫描方式:采用选择性离子监测(SIM)采集;α-氯甲苯的特征离子为m/z 91、126、65,选择不同的离子通道,以m/z 91作为定量离子,以m/z 91、126、65作为定性鉴别离子,考察各特征离子与m/z 91离子的丰度比。
在以上色谱、质谱条件下,α-氯甲苯的特征离子见表2。
表2 待测成分的定性离子
待测成分分子式特征选择离子及丰度比
α-氯甲苯C6H5CH2Cl 91(100),126(26),65(13)
6 计算
w(被测成分)= ρV/m
式中:w(被测成分)——样品中被测成分的质量分数,μg/g;
——从标准曲线中得出的待测溶液中被测成分的质量浓度,μg/mL;
V ——样品定容体积,mL;
m ——样品取样量,g。
7 回收率和精密度
多家实验室验证低浓度的平均提取回收率在91.9%~104.5%之间,相对标准偏差小于3.8%(n=6);高浓度的平均提取回收率在92.6%~107.8%之间,并且RSD小于3.2%(n=6)。
8 色谱图、质谱图
图1 α-氯甲苯标准溶液的气相色谱图(α-氯甲苯TR=7.340min)
图2 α-氯甲苯的质谱图。