双酶切编辑
载体双酶切

载体双酶切技术是分子生物学领域常用的实验技术之一,它通过利用两种不同的限制性内切酶作用于同一个质粒DNA,从而实现对目标DNA片段的精确剪切和克隆。
本文将从载体双酶切技术的原理、应用和优缺点等方面进行详细介绍。
一、原理载体双酶切技术的原理主要基于两种不同的限制性内切酶对DNA的特异性切割。
首先,选择两种能够在同一质粒上切割出相互兼容的黏性末端的内切酶,并确定其最佳反应条件。
然后,将目标DNA片段与质粒进行双酶切反应,使得目标DNA片段的末端与质粒的末端具有互补的黏性末端。
最后,通过DNA连接酶的作用,将目标DNA片段与质粒连接成重组质粒,实现目标DNA片段的克隆插入。
二、应用载体双酶切技术在分子生物学研究中有着广泛的应用。
首先,它可以用于重组质粒的构建,将外源基因插入到质粒中,用于基因克隆、表达和功能研究。
其次,还可以通过双酶切技术对质粒进行定向修饰,如引入点突变、插入序列或者删除特定片段等,用于研究基因的结构与功能。
此外,载体双酶切技术也被广泛应用于基因工程、蛋白质表达、基因组编辑等领域。
三、优缺点1. 优点(1)精准:通过双酶切技术可实现对DNA片段的精确切割和定向连接,保证了重组质粒的稳定性和可靠性。
(2)灵活:可以根据实验需要选择不同的限制性内切酶组合,实现对DNA的多样化操作。
(3)高效:相比传统的单酶切技术,载体双酶切技术能够提高DNA片段的连接效率和克隆成功率。
2. 缺点(1)操作复杂:双酶切技术需要充分考虑两种内切酶的选择、反应条件的优化及连接方法的调整,操作过程较为复杂。
(2)局限性:某些情况下可能无法找到适合的双酶切位点,导致目标DNA片段无法有效插入到质粒中。
(3)成本较高:需要购买多种限制性内切酶和连接酶,增加了实验成本。
综上所述,载体双酶切技术作为一种重要的分子生物学工具,在基因工程和分子生物学研究中发挥着重要作用。
随着技术的不断进步和完善,相信它将在更多领域展现出其巨大的潜力和价值。
双酶切实验

双酶切概述双酶切反应(Double Digests)1、同步双酶切同步双酶切是一种省时省力的常用方法。
选择能让两种酶同时作用的最佳缓冲液是非常重要的一步。
NEB每一种酶都随酶提供相应的最佳NEBuffer,以保证100%的酶活性。
NEBuffer的组成及内切酶在不同缓冲液中的活性见《内切酶在不同缓冲液里的活性表》及每支酶的说明书。
能在最大程度上保证两种酶活性的缓冲液即可用于双酶切。
由于内切酶在非最佳缓冲液条件下的切割速率会减缓,因此使用时可根据每种酶在非最优缓冲液中的具体活性相应调整酶量和反应时间。
2、分步酶切如果找不到一种可以同时适合两种酶的缓冲液,就只能采用分步酶切。
分步酶切应从反应要求盐浓度低的酶开始,酶切完毕后再调整盐浓度直至满足第二种酶的要求,然后加入第二种酶完成双酶切反应。
3、使用配有特殊缓冲液的酶进行双酶切(图)使用配有特殊缓冲液的酶进行双酶切也不复杂。
在大多数情况下,采用标准缓冲液的酶也能在这些特殊缓冲液中进行酶切。
这保证了对缓冲液有特殊要求的酶也能良好工作。
由于内切酶在非最佳缓冲液中进行酶切反应时,反应速度会减缓,因此需要增加酶量或延长反应时间。
通过《内切酶在不同缓冲液里的活性表》可查看第二种酶在特殊缓冲液相应盐浓度下的作用活性。
双酶切建议缓冲液注:只要其中一种酶需要添加BSA,则应在双酶切反应体系中加入BSA。
BSA不会影响任何内切酶的活性。
注意将甘油的终浓度控制在10%以下,以避免出现星号活性,详见《星号活性》。
可通过增加反应体系的总体积的方法实现这一要求。
某些内切酶的组合不能采用同步双酶切法,只能采用分步法进行双酶切。
上表中这些组合以“se q”标注。
[编辑本段]双酶切的注意事项1、做转化的时候,进行酶连接反应时,注意保持低温状态,因为LIGASE酶很容易降解.为保险起见,一般连接3小时,16度。
2、对含有AMP-RESISTENCE的质粒铺板时,注意加AMP时的温度,温度过高,会使克隆株无法筛选出来.我的方法是培基高温消毒后放在烤箱里,烤箱一般温度为55-60度,然后做的时候拿出来,这样好掌握温度。
Oligo 7操作方法
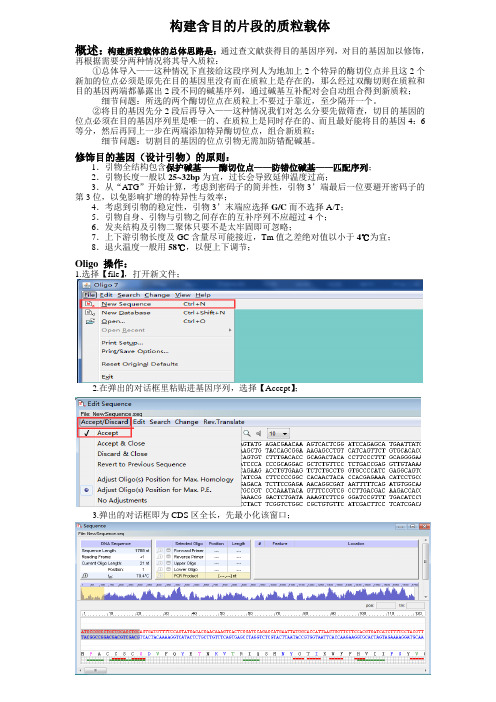
构建含目的片段的质粒载体概述:构建质粒载体的总体思路是:通过查文献获得目的基因序列,对目的基因加以修饰,再根据需要分两种情况将其导入质粒:①总体导入——这种情况下直接给这段序列人为地加上2个特异的酶切位点并且这2个新加的位点必须是原先在目的基因里没有而在质粒上是存在的,那么经过双酶切则在质粒和目的基因两端都暴露出2段不同的碱基序列,通过碱基互补配对会自动组合得到新质粒;细节问题:所选的两个酶切位点在质粒上不要过于靠近,至少隔开一个。
②将目的基因先分2段后再导入——这种情况我们对怎么分要先做筛查,切目的基因的位点必须在目的基因序列里是唯一的、在质粒上是同时存在的、而且最好能将目的基因4:6等分,然后再同上一步在两端添加特异酶切位点,组合新质粒;细节问题:切割目的基因的位点引物无需加防错配碱基。
修饰目的基因(设计引物)的原则:1.引物全结构包含保护碱基——酶切位点——防错位碱基——匹配序列;2.引物长度一般以25~32bp为宜,过长会导致延伸温度过高;3.从“ATG”开始计算,考虑到密码子的简并性,引物3’端最后一位要避开密码子的第3位,以免影响扩增的特异性与效率;4.考虑到引物的稳定性,引物3’末端应选择G/C而不选择A/T;5.引物自身、引物与引物之间存在的互补序列不应超过4个;6.发夹结构及引物二聚体只要不是太牢固即可忽略;7.上下游引物长度及GC含量尽可能接近,Tm值之差绝对值以小于4℃为宜;8.退火温度一般用58℃,以便上下调节;Oligo 操作:1.选择【file】,打开新文件;2.在弹出的对话框里粘贴进基因序列,选择【Accept】;3.弹出的对话框即为CDS区全长,先最小化该窗口;4.选择【edit】——【forward primer】,编辑上游引物;4.从全长序列5’端复制前30个碱基粘贴进新弹出的对话框中编辑,第一步筛选酶切位点【search】—【for Restriction Sites】,主要看是否是多克隆位点判断是否为多克隆位点(②法导入须看)提示目的基因里没有的酶切位点(①法导入须看)5.配合质粒图筛选出合适的两端酶切位点(质粒中存在而目的基因里不存在)6.在第4步得到的30位匹配序列对话框中,参照图5输入酶切位点序列→参照附录2输入保护碱基→参照原则输入防错位碱基,再在尾端删除配对序列碱基直至符合原则要求;7.上游引物就设计好了,点击确定保存,跳到全序列窗口,点击【change】--【Strands】转向反向互补链,复制30个,同理编辑下游引物;8.点击确定9.点击save,保存结束。
双酶切体系梳理

Double Digestion(双酶切反应)时Universal Buffer(通用缓冲液)的使用表■ 说明使用二种酶同时进行DNA切断反应(Double Digestion) 时,为了节省反应时间,通常希望在同一反应体系内进行。
TaKaRa采用Universal Buffer表示系统,并对每种酶表示了在各Universal Buffer中的相对活性。
尽管如此,在进行Double Digestion时,有时还会难以找到合适的Universal Buffer。
本表以在pUC系列载体的多克隆位点处的各限制酶为核心,显示了在Double Digestion可使用的最佳Universal Buffer条件。
在本表中,各Universal Buffer 之前表示的[数字×] 是指各Universal Buffer的反应体系中的最终浓度。
TaKaRa销售产品中添附的Universal Buffer全为10倍浓度的缓冲液。
终浓度为0.5×时反应体系中的缓冲液则稀释至20倍,1×时稀释至10倍,2×时稀释至5倍进行使用。
■ 注意◇1 μg DNA中添加10 U的限制酶,在50 μl的反应体系中,37℃下反应1小时可以完全降解DNA。
◇为防止Star活性的产生,请将反应体系中的甘油含量,尽量控制在10%以下。
◇根据DNA的种类,各DNA的立体结构的差别,或当限制酶识别位点邻接时,有时会发生Double Digestion不能顺利进行的可能。
(精选文档,可编辑word,整理文档不易,建议收藏) (精选文档,可编辑word,整理文档不易,建议收藏) (精选文档,可编辑word,整理文档不易,建议收藏) (精选文档,可编辑word,整理文档不易,建议收藏) (精选文档,可编辑word,整理文档不易,建议收藏) (精选文档,可编辑word,整理文档不易,建议收藏) (精选文档,可编辑word,整理文档不易,建议收藏) (精选文档,可编辑word,整理文档不易,建议收藏) (精选文档,可编辑word,整理文档不易,建议收藏) (精选文档,可编辑word,整理文档不易,建议收藏)。
生物技术中的基因编辑疗法

生物技术中的基因编辑疗法近年来,随着生物技术的发展,基因编辑疗法成为了治疗基因突变疾病的新方法。
基因编辑疗法是指通过人工干预基因序列,使得有缺陷或异常的基因得以正常表达,从而达到治疗目的的方法。
一、基因编辑疗法的原理基因编辑疗法的原理是通过利用已知基因序列的信息来进行人工操作,使得有害的基因突变被改变成健康的基因状态。
它的操作主要分为三个步骤: “切”,“改”和“贴”。
“切”是指利用特定酶切割DNA,使得需要编辑的基因部位的序列被切开。
这些酶被称为核酸酶,它们能将连接在一起的DNA双链裂解开来,同时可以选择性地切割特定的序列。
“改”指的是在切割DNA后,通过在对应的位点上插入外源性DNA或RNA序列,最终改变基因的状态。
这种修改基因序列的方式被称为转录翻译,是一种非常有效且广泛应用的方法。
“贴”是指在修改基因序列后,再通过基因修复酶将基因序列恢复成完整的状态,从而达到修复因基因突变而导致的疾病的目的。
二、基因编辑疗法的治疗现状基因编辑疗法现在已经应用于一些少见的遗传性疾病,例如囊性纤维化、苯丙酮尿症等。
这些疾病的发生与特定基因的缺陷或突变有关,通过基因编辑可以达到恢复这些基因的正常状态,从而达到治疗疾病的目的。
另外,在癌症的治疗中也可以应用基因编辑疗法。
基因编辑技术可以通过删除癌细胞中的特定基因,使得癌细胞失去生存能力,从而达到治疗目的。
尽管现在癌症的治疗复杂度很高,但基因编辑疗法仍有很大的发展空间。
三、基因编辑疗法的未来发展随着基因编辑技术的不断发展,基因编辑疗法也将会得到迅速的发展。
未来,基因编辑疗法将会出现更多的应用场景,例如针对心血管疾病、神经系统疾病等疾病的治疗。
基因编辑疗法的发展将会改变现在的医学模式,并且会对整个医学领域产生深远的影响。
但是,由于技术的限制和道德伦理的问题,基因编辑疗法的应用和推广仍需要一个良好的环境和政策的支持。
四、基因编辑疗法的风险基因编辑疗法主要存在以下风险:(1)风险1:基因编辑会引入新的变异,导致更大的健康问题。
基因编辑技术

P-AATTCGT OH-GCA
-ACGAATTCGT-TGCTTAAGCA
3、DNA聚合酶
具有5`→3`聚合活性、5`→3`外切酶活性、3`→5`外 切酶活性。
可用于合成双链cDNA分子或片断连接,探针制备,序 列分析等。
耐热DNA聚合酶 最适反应温度为75~80℃ 具有5ˊ→3ˊ外切酶活性,不具有3ˊ→5ˊ外切酶活性
构建与靶基因同源 (10~15kb)的载体
技 外显子插有新霉素抗
术 性基因 (neor)作为
路
正选择
疱疹病毒胸苷激酶 (HSV-tk)基因作为
负选择
线 将重组体导入ES细胞,体外培养
neor 和HSV-tk作双重筛选 存活的ES细胞
显微注射
回交得到X被敲除的动物
表型分析
图 示
ZFN 技术原理
锌指核酸酶(Zinc-finger nucleases, ZFN)是人工改造的限制性核酸内 切酶,利用不同的锌指结构识别特异DNA序列,利用核酸酶切断靶DNA。
第四节 重组DNA技术常用载体
一、定义
是指能携带目的基因进入宿主细胞进行扩增和表达的 一类DNA分子。
分类: 克隆载体:用于在宿主细胞中隆和扩增外
源DNA片段。 表达载体:用于在宿主细胞中获得外源基因
表达产物。
二、特点
1、具备复制能力。 2、具备一个或多个筛选标志。 3、具备较多拷贝数,易从宿主细胞中分离纯化。 4、具备一个或多个限制性内切酶的单一识别点(多
克隆位 点,MCS)。
5、具有较高的遗传稳定性。
3、常用载体
质粒DNA、噬菌体DNA、病毒DNA、酵母DNA
这些载体均需经人工构建,除去致病基因,并赋 予一些新的功能:如有利于进行筛选的标志基因、单一 的限制酶切点等。
双酶切载体构建法

双酶切载体构建法双酶切载体构建法是一种常用的分子生物学技术,用于构建重组DNA。
在这种方法中,通过利用两个特定的限制性内切酶,将目标DNA分子切割,并将其插入到载体DNA中,从而构建重组DNA。
本文将详细介绍双酶切载体构建法的原理、步骤和应用。
一、双酶切载体构建法的原理双酶切载体构建法的原理基于限制性内切酶的作用。
限制性内切酶是一类能够识别并切割特定DNA序列的酶。
在双酶切载体构建法中,我们选择两个限制性内切酶,一个用于切割目标DNA,另一个用于切割载体DNA。
这两个酶的切割位点必须位于不同的位置,以确保目标DNA能够正确地插入到载体DNA中。
二、双酶切载体构建法的步骤1. 选择限制性内切酶:首先,根据目标DNA的序列,选择能够识别并切割目标DNA的限制性内切酶。
同时,选择一个在载体DNA 中有适当切割位点的限制性内切酶。
2. 切割目标DNA和载体DNA:将目标DNA和载体DNA分别与两个限制性内切酶一起反应。
这样,目标DNA和载体DNA就会在限制性内切酶的作用下被切割成多个片段。
3. 准备载体DNA:将载体DNA经过限制性内切酶切割后,使用凝胶电泳等方法,将切割后的载体DNA片段分离出来。
4. 连接目标DNA和载体DNA:将切割后的目标DNA与载体DNA片段在适当的条件下进行连接。
这可以使用DNA连接酶来完成,它能够将两个DNA片段连接在一起。
5. 转化宿主细胞:将连接好的重组DNA导入宿主细胞中。
这可以通过转染、电穿孔等方法完成。
6. 筛选和鉴定重组DNA:在转化宿主细胞后,使用选择性培养基或荧光筛选等方法,筛选出携带重组DNA的细胞。
然后,通过PCR、限制性酶切、测序等方法,对重组DNA进行鉴定和验证。
三、双酶切载体构建法的应用双酶切载体构建法在分子生物学研究中有广泛的应用。
以下是它的几个主要应用领域:1. 基因克隆:双酶切载体构建法可以用于将外源基因插入到载体DNA中,从而构建重组DNA,进而实现外源基因的克隆。
《双酶切及连接》课件

• 双酶切技术简介 • 双酶切的实验步骤 • 双酶切的应用 • 双酶切的注意事项 • 双酶切技术的发展趋势
01
双酶切技术简介
酶切技术的定义
01
02
03
酶切技术定义
酶切技术是一种利用酶的 专一性对特定底物进行切 割的生物技术。
酶的专一性
酶只对特定的底物起作用 ,切割位点具有高度专一 性。
双酶切技术的改进与创新
新型限制性核酸内切酶的 开发
随着生物技术的不断发展,新型限制性核酸 内切酶不断涌现,为双酶切技术提供更多选 择和灵活性。
自动化双酶切系统的研发
通过自动化技术实现双酶切的快速、高效和 标准化操作,提高实验效率并减少人为误差
。
双酶切技术的发展前景
01
双酶切技术在基因克隆和基因治 疗等领域的应用前景广阔,未来 将继续发挥重要作用。
05
双酶切技术的发展趋势
双酶切与其他技术的结合
双酶切与PCR技术的结合
通过双酶切技术将目的基因和载体进行酶切,再利用PCR技术进行扩增和鉴定,提高基 因克隆的效率和准确性。
双酶切与基因编辑技术的结合
将双酶切技术与CRISPR-Cas9等基因编辑技术结合,实现对特定基因的敲除、敲入和 定点突变等操作,为基因功能研究和基因治疗提供有力工具。
02
随着生物技术的不断进步,双酶 切技术将与其他技术不断融合创 新,为生命科学研究提供更多有 力工具。
THANKS
感谢观看
酶切技术的分类
单酶切技术
使用一种限制性内切核酸酶对 DNA进行切割。
双酶切技术
使用两种不同的限制性内切核酸酶 对DNA进行切割,通常用于产生 具有不同黏性末端的DNA片段, 便于后续的连接反应。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
2连接反应
3注意事项
1简介编辑双酶切反应(Double Digests)
1、同步双酶切
同步双酶切是一种省时省力的常用方法。选择能让两种酶同时作用的最佳缓冲液是非常重要的一步。NEB每一种酶都随酶提供相应的最佳NEBuffer,以保证100%的酶活性。NEBuffer的组成及内切酶在不同缓冲液中的活性见《内切酶在不同缓冲液里的活性表》及每支酶的说明书。能在最大程度上保证两种酶活性的缓冲液即可用于双酶切。由于内切酶在非最佳缓冲液条件下的切割速率会减缓,因此使用时可根据每种酶在非最优缓冲液中的具体活性相应调整酶量和反应时间。
双酶切建议缓冲液
注:
只要其中一种酶需要添加BSA,则应在双酶切反应体系中加入BSA。BSA不会影响任何内切酶的活性。
注意将甘油的终浓度控制在10%以下,以避免出现星号活性,详见《星号活性》。可通过增加反应体系的总体积的方法实现这一要求。
某些内切酶的组合不能采用同步双酶切法,只能采用分步法进行双酶切。上表中这些组合以“seq”标注。
3、转化:
a、一般转化仅需要加入2μl加入至100μl正常的TOP10感受态细胞中,冰浴放置30分钟。
b、再在水浴中42℃热激一般90~120秒钟后,再在冰中放置3分钟。
c、加入800μl无抗生素培养基,37℃全温振荡摇床培养40分钟。
取100μl涂布平板。一般转化质粒不建议离心涂布(除非感受态效价特别低),
双酶切编辑
做转化的时候,进行酶连接反应时,注意保持低温状态,因为LIGASE酶很容易降解。为保险起见,一般连接3小时,16度;对含有AMP-RESISTENCE的质粒铺板时,注意加AMP时的温度,温度过高,会使克隆株无法筛选出来。我的方法是培基高温消毒后放在烤箱里,烤箱一般温度为55-60度,然后做的时候拿出来,这样好掌握温度。铺板前后注意用吹风机吹干;对照的设立:为验证双酶切是否成功。
酶量的问题:以TAKARA的为例,其对1单位酶的定义如下:在50μl反应液中,37℃温度下反应1小时,将1μg的λDNA完全分解的酶量定义为1个活性单位(U)。而该酶浓度约为15单位/微升,在除外酶降解的因素外,该酶可分解15μg的DNA,而一般从1-4ml菌液提出的DNA约为3μg,而PCR纯化后的产物(50体系)约为3μg,所以即便全部加进去,只要纯化的质量好,酶切完全切得动。
2、酶切、回收后的PCR产物与载体的连接
摩尔比的计算,很多人凭经验也可以。但对于初学者从头认真计算则非常有必要。回收的载体片段:回收的PCR产物片段=1:3到1:10,一般取前者0.03pmol,后者取0.3pmol。
pmol为单位的DNA转换为为μg单位的DNA:(X pmoles×长度bp×650)/ 1,000,000(注:长度bp×650是该双链DNA的分子量)所得数值即为μg,也可以直接用这个公式套.1pmol 1000bp DNA=0.66μg,如载体是5380bp,则0.03pmol为
经PCR鉴定,克隆90%-100%的阳性率,所以在后面的挑克隆中,我只挑选4个就足够了。然后双酶切鉴定,测序。
3注意事项编辑1、做转化的时候,进行酶连接反应时,注意保持低温状态,因为LIGASE酶很容易降解.为保险起见,一般连接3小时,16度。
2、对含有AMP-RESISTENCE的质粒铺板时,注意加AMP时的温度,温度过高,会使克隆株无法筛选出来.我的方法是培基高温消毒后放在烤箱里,烤箱一般温度为55-60度,然后做的时候拿出来,这样好掌握温度。铺板前后注意用吹风机吹干。
几个非常重要的问题
1做转化质粒的时候一般不加连接酶。对PCR产物进行酶连接反应时,注意保持低温状态,因为T4连接酶长时间放置在常温下容易活性降低,建议在冰盒中操作。
2对含有AMP-RESISTENCE的质粒涂布时,一般培养基会放置在培养箱中一段时间后才会到感受态中,
3对照的设立:
为验证双酶切是否成功,可做如下对照:
创建者:1袁泉2
词条贡献榜
突出贡献者:
干活禁言
百科消息:
“二维马”知识行大挑战
4.19激情秒杀五星级酒店
足不出户看三星堆博物馆
商城许愿树:大声说出你的爱
百科新玩法:新版城市百科上线
推广链接
赛默飞FastDigest快速限..
门研发的进..
2、分步酶切
如果找不到一种可以同时适合两种酶的缓冲液,就只能采用分步酶切。分步酶切应从反应要求盐浓度低的酶开始,酶切完毕后再调整盐浓度直至满足第二种酶的要求,然后加入第二种酶完成双酶切反应。
3、使用配有特殊缓冲液的酶进行双酶切(图)
使用配有特殊缓冲液的酶进行双酶切也不复杂。在大多数情况下,采用标准缓冲液的酶也能在这些特殊缓冲液中进行酶切。这保证了对缓冲液有特殊要求的酶也能良好工作。由于内切酶在非最佳缓冲液中进行酶切反应时,反应速度会减缓,因此需要增加酶量或延长反应时间。通过《内切酶在不同缓冲液里的活性表》可查看第二种酶在特殊缓冲液相应盐浓度下的作用活性。
B.酶切过的未进行连接反应的双酶切产物,进行转化,这一步可以证明是否有残留的未被任何酶切的原始质粒
C.设原始质粒为对照,意为检测整个操作过程中是否有误.
D.AMP阴性板上用同一批感受态细胞铺板20微升足够,检测感受态状况.
4.所有的试剂切记低温保存.一步一个脚印.不要偷懒,图省事最后却更费事.注意设立对照。
#Enzyme
分享到:
举报浏览(1094)评论转载
你可能也喜欢
乐高块砖做成的汽车
【脚踏版法拉利】
【空间周刊 - 小编风采 - 第6期】特别访谈:水冰月(娜娜)
2连接反应编辑1、回收PCR产物:
在进行PCR扩增时候,给引物两端设计好酶切位点,一般说来,限制酶的选择非常重要,尽量选择粘端酶切和那些酶切效率高的限制酶,如BamHI,HindIII,提前看好各公司的双切酶所用公用的BUFFER,以及各酶在公用BUFFER里的效率。选好酶切位点后,在各个酶的两边加上保护碱基。
3、对照的设立:为验证双酶切是否成功,可做如下对照:
酶切反应时加各单酶分别切,两管,用同一种BUFFER,跑胶,看单切的两管是否成线性.如两管均成线性可初步判断双酶切成功.做转化时,也要进行对照。
词条标签:生物酶切
双酶切图册
词条统计
浏览次数:46757次
编辑次数:12次历史版本
最近更新:2013-07-17
双酶切时间及其体系:需要强调的是很多人建议酶切过夜,其实完全没有必要,我一般酶切3个小时,其实1个小时已经足够。应用大体系,如100微升。
纯化问题:纯化PCR产物割胶还是柱式,我推荐柱式,因为割胶手法不准,很容易割下大块的胶,影响纯化效率。现在的柱式纯化号称可以祛除引物,既然如此,酶切掉的几个碱基肯定也会被纯化掉了。所以,PCR产物和双酶切产物的纯化均可应用柱式纯化。我用的是TAKARA的纯化柱试剂盒
0.03×5.38×0.66=0.106524μg。
测DNA浓度测定可使用微量可见光分光光度计或者可以使用艾本德的BioPhotometer核酸蛋白分析仪,注意OD值,一般约1.7-1.9.的说明写的过夜,而其对连接酶单位的定义为:在20μl的连接反应体系中,6μg的λDNA-Hind III的分解物在16℃下反应30分钟时,有90%以上的DNa段被连接所需要的酶量定义为1个活性单位(U)。而它的浓度为350 U/μl,所以完全够用。连接酶容易失活,注意低温操作,最好在冰上。时间3个小时足已。
双酶切的buffer选择
如果使用NEB的内切酶,查询双酶切buffer请点击这里。
注:
只要其中一种酶需要添加BSA,则应在双酶切反应体系中加入BSA。BSA不会影响任何内切酶的活性。
注意将甘油的终浓度控制在10%以下,以避免出现星号活性,可通过增加反应体系的总体积的方法实现这一要求。
某些内切酶的组合不能采用同步双酶切法,只能采用分步法进行双酶切。上表中这些组合以"seq"标注。
A酶切反应时加各单酶分别切,两管,用同一种BUFFER,跑胶,看单切的两管是否成线性.如两管均成线性可初步判断双酶切成功.
做转化时,也要进行对照.设4个:
A.即拿双酶切的质粒产物也进行连接反应,这个对照可进一步看双酶切是否成功,如果长出克隆,说明很有可能只进行了单酶切,如没长出克隆,则证明双酶切成功,当然要保证感受态,培基,连接酶都'正常'的情况下.