ISO13485受控文件清单
ISO13485质量记录清单

B/0
5年
49
QR-QP8.3-01
《不合格处置单》
B/0
5年
50
QR-QP8.3-02
《退换货申请表》
B/0
5年
51
QR-QP8.3-03
《不合格品销毁登记表》
B/0
5年
52
QR-QP8.5-01
《纠正预防措施处理单》
B/0
5年
53
QR-QP8.5.1-01
《忠告性通知发布记录》
B/0
5年
54
QR-AD01-01
《固定资产入库单》
B/0
5年
55
QR-AD01-02
《固定资产出库单》
B/0
5年
56
QR-AD01-03
《固定资产报废审批单》
B/0
5年
57
QR-AD01-04
《固定资产内部调拨单》
B/0
5年
58
QR-AD01-05
《固定资产借用登记单》
B/0
5年
59
QR-AD01-06
《监视测量设备检定记录》
B/0
5年
39
QR-QP8.2.1-01
《顾客满意程度调查表》
B/0
5年
40
QR-QP8.2.1-02
《顾客投诉记录》
B/0
5年
41
QR-QP8.2.3-01
《可疑医疗器械不良事件报告表》
B/0
5年
42
QR-QP8.2.3-02
《医疗器械不良事件43
QR-QP8.2.3-03
QR-QP7.4-01
《供应商名录》
ISO13485记录控制程序(含表格)

记录控制程序(ISO13485-2016/YYT0287-2017)1.0目的作为质量管理体系运行有限运行证据,保证产品可追溯性,为持续改进提供证据支持。
2.0范围适用于公司质量记录和报告等的管理。
3.0引用/参考文件ISO9001:2015YY/T0287-2017/ISO13485:2016医疗器械GMP无菌医疗器械实施细则《文件与资料控制程序》4.0职责4.1质量部负责公司所有质量记录和报告等的控制与管理。
4.2各职能部门负责本部门质量记录的控制与管理。
5.0作业程序5.1记录5.1.1批记录:批生产记录、批包装记录和批检验记录。
5.1.2通用记录:流程表单、台帐、标识卡、管理卡等。
5.2记录编制5.2.1记录由使用部门组织人员根据管理标准和技术标准进行设计,纳入对应的文件作为附件,按照《文件与资料控制程序》规定的权限职责要求进行审批。
5.2.2记录标题要明确表达记录的意义、性质并有独一无二的识别编号,记录编号规则参照《文件与资料控制程序》;记录内容要详尽、合乎逻辑,符合ISO 和GMP的要求,要包括所有必要的内容。
5.2.3记录中的操作指令、步骤、参数及引用的文件编号是对产品技术特性及质量特性的阐述和指导,因此应达到如下要求:5.2.3.1术语规范、数据准确、无误,符合法定标准文件。
5.2.3.2语言要精炼、明确,项目要清晰,符合公司标准的要求。
5.2.3.3记录的格式要符合ISO和GMP的要求,并结合企业生产管理和质量监控的实际要求来编制,要提供足够的空白格,以便于填写。
5.2.3.4设计记录时,要尽量考虑到如何有效地防止填写错误或差错。
5.2.3.5各种技术、质量参数和技术经济指标度量单位均按国家计量法规的规定采用国际标准计量单位执行。
5.3记录填写5.3.1记录只能由规定的人员及时填写,不允许事前先填或事后补填。
5.3.2填写内容应保持绝对的真实,字迹清晰、工整,统一使用黑色签字笔填写。
商贸企业质量管理 程序文件(模板) ISO13485 通用版本
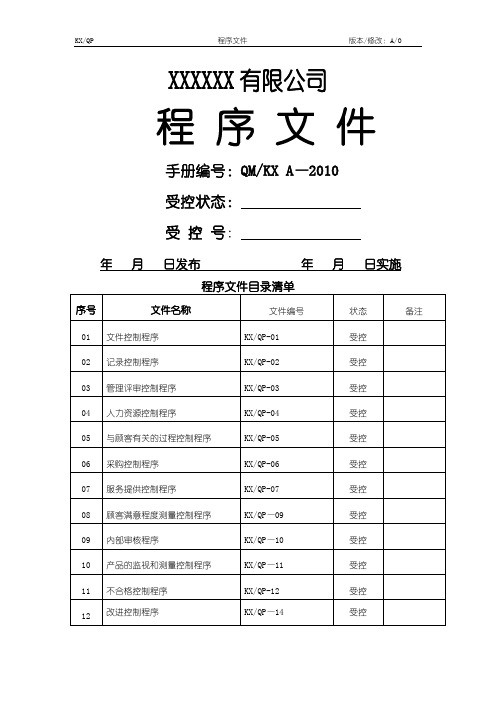
XXXXXX有限公司程序文件手册编号:QM/KX A—2010受控状态:受控号:年月日发布年月日实施程序文件目录清单4.2.3文件控制程序文件编号:KX/QP—011目的对与本公司质量管理体系有关的文件进行控制,确保各相关场所和部门获得适用文件的有效版本。
2适用范围适用于所有与质量管理体系有关的文件控制.3职责3.1总经理负责批准发布《管理手册》;3。
2管理者代表负责组织编写、审核《管理手册》,批准发布二级文件;3.3办公室负责质量管理体系文件的发放,以及对现有质量管理体系文件进行定期评审;3。
4各个部门负责相关二级文件的编审、使用与质量管理体系有关的文件的收集、整理并交办公室归档保存。
4内容与要求4。
1文件管理工作流程4.2文件分类与保管4.2.1管理手册(包含了所有过程的程序文件),由办公室备案保存,持有人在使用中注意保管。
4.2。
2公司二级文件分为两类:a)部门工作手册,作为各个部门运行质量管理体系的日常实施细则,由使用部门保存,办公室备案存档。
b)其他质量文件,包括针对特定产品、项目或合同编制的质量计划、服务标准、销售文件和质量目标策划、质量管理体系策划等,文件的组成应适合于其特有的活动方式,由各个相关的责任部门保存、使用,并报办公室存档备查。
4。
2。
3公司管理性文件,如各种行政管理制度、部分外来管理性文件,包括与质量管理体系有关的法律法规文件等,由办公室按本程序相关条款执行.4.3文件的编号4.3.1质量管理体系文件的编号:a)管理手册编号说明公司名称代号—管理手册代号—版次/修改状态,手册中各章节以章节号区分。
例如:QM/KX—,表示指石首康鑫商贸有限公司管理手册.KX/QP-01表示指石首康鑫商贸有限公司第1个程序文件b)各部门三级文件编号说明:公司名称代号- /部门代号-文件顺序号.例如:KX/XS-01,表示销售部第1号文件。
4。
4文件的编写、审核、批准、发放文件发布前应得到批准,以确保文件是适宜的:4.4.1管理手册由贯标工作小组负责编写,管理者代表审核,上报总经理批准发布,办公室做好发放登记。
ISO13485记录表格 汇总

地址
邮政编码
产品/类别
电话/传真
联系人
质量得分(占60%):(合格批次/到货总批次)×60
质量评分:
交货期(占20%)(按期到货的批次/到货总批次)×20
质量评分:
其他(占20%)(包括价格、包装、售后服务等)
质量评分:
总评分及处理建议:
生产供应部负责人: 日期:
主管领导意见:
签名: 日期:
性能指标
随机附件
和文件
维修记录
备注
设备封存(启用)申请表
序号:编号:GNKJ/QR-6.3-09
设施名称
设备编号
型号/规格
申请日期
使用部门
封存/启用
主要技术性能:
封存/启用说明:
备注:
生产供应部意见:
经理:
质量管理部意见:
经理:
技术部意见:
经理:
主管领导批准:申请人:
低值易耗品报废单
序号:编号:GNKJ/QR-6.3-10
供方现场评定记录
序号:编号:GNKJ/QR-7.4-06
供方名称
供品名称
电话/传真
联系人
地址
邮政编码
设备能力:
评定结论:
评定人员:职务:
日期:
人员能力:
检测能力:
体系能力:
记录:审核;批准:
物资采购合同评审表
序号:表格编号:GNKJ/QR-7.4-07
合同类别
采购合同编号
供方名称
供方营业执照号
采购物资
序号
文件名称
编号
版本/状态
复印份数
复印人
领导审批
复印时间
备注
医疗器械ISO13485质量手册、程序文件、表单汇编---乐得施

质量手册(包括:程序文件、表单全套)依据: ISO13485:2003、YY/T0287—2003、YY/T0316—2003编制:审核:批准:2014-09-28发布2014—10—01实施广州乐得施医疗用品有限公司目录序号标题编页1 目录2—32 质量手册发布令3 任命书4 主题内容75 公司概况8-106 目的范围117 质量方针质量目标质量承诺128 组织机构139 质量管理体系职责分配表1410 质量管理体系15—1611 文件控制程序17-1912 记录控制程序20-2113 管理职责22-2614 管理评审控制程序27—2815 资源管理控制程序29-3116 产品实现3217 产品实现的策划控制程序33-3418 与顾客有关的过程控制程序35-3719 采购控制程序38-4020 服务控制程序41—4521 监视和测量装置控制程序46-4722 顾客满意度测量控制程序48-4923 内部审核控制程序50-5324 过程和产品的监视与测量控制程序54—5525 不合格品控制程序56—5826 数据分析控制程序59—6027 纠正、预防和改进措施控制程序61—6228 医疗器械经营企业质量管理全套表格63-124ISO13485质量管理全套表格目录1.首营企业审批表2.首营品种审批表3.温湿度记录表4.质量问题跟踪表5.产品质量投诉处理记录6.2014年度员工培训记录7.不合格品处理记录表8.不良事件报告记录9.医疗器械质量事故调查报告10.医疗器械质量事故统计表11.程序文件执行情况自查情况表12.不良事件报告记录13.厂区环境卫生检查记录表14.车间门窗墙壁天花板清洁记录15.设施和设备安装、维修、调试及定期检查、保养记录16.医疗器械购进、验收、入库记录17.出库单18.入库单19.产品出库、复核、销售记录20.商品投诉、质量查询报告单21.医疗器械商品养护记录22.医疗器械产品出库、复核记录23.医疗器械产品购进记录24.医疗器械产品销售记录25.医疗器械产品验收/检验记录26.商品投诉、质量查询记录27.医疗器械产品养护、检查记录28.医疗器械售后服务反馈登记表29.医疗器械效期产品管理记录30.用户访问联系记录表31.售后服务登记表32.医疗器械售后服务反馈登记表33.医疗器械销售产品召回记录34.事故初始报告和最终报告书35.设计开发表格汇编36.医疗器械风险管理计划37.医疗器械风险管理报告38.风险评价、风险控制措施记录39.表生产和生产后信息评价和处理记录40.产品安全特征问题清单质量手册发布令依据YY/T0287-2003标准要求,结合本公司产品特性,在管理者代表的组织下,通过对企业质量管理体系的策划和建立,编制了本质量手册(包括程序文件),现予以发布实施。
最新医疗防护用品ISO13485:2016一整套程序文件含表单

适用于与质量体系有关的文件控制管理。 3. 职责 3.1 总经理负责质量方针与质量目标的制定与批准;负责质量手册的批准。 3.2 管理者代表负责各部门质量目标、程序文件的批准及程序文件发放范围的批 准;负责组织对质量手册和程序文件的评审。 3.3 行政部负责文件的发放、回收、销毁,原稿的保存(包括电子版)及做好相关 记录;负责编制并及时更新《文件发放总览表》;负责法律法规与标准及外来执行 文件的控制。 3.4 各部门负责相关文件的编制和使用保管。部门经理负责制定本部门质量目标; 批准三阶文件;组织对三阶文件及部门其它文件的评审。 4. 定义 4.1 质量体系文件
2020.01.02 2020.01.02 2020.01.02 2020.01.02 2020.01.02 2020.01.02 2020.01.02 2020.01.02
2020.01.02 2020.01.02 2020.01.02 2020.01.02 2020.01.02 2020.01.02 2020.01.02 2020.01.02 2020.01.02 2020.01.02 2020.01.02 2020.01.02 2020.01.02 2020.01.02 2020.01.02 2020.01.02 2020.01.02
34
产品放行控制程序
二、 配套表单
QP-32 QP-33 QP-34
2020.01.02
A/0
2020.01.02
A/0
2020.01.02
A/0
XXXXX 有限公司 程序文件
文件编号 版号
QP-01 A.0
文件管理程序
页次
1/5
生效日期 2020/01/02
ISO13485文件与资料控制程序
文件与资料控制程序(ISO13485-2016/YYT0287-2017)1.0目的对公司质量管理体系所要求的文件与资料进行控制,确保各相关场所使用的文件与记录受控。
2.0适用范围适用于公司与质量管理体系有关的所有文件与资料的控制。
3.0引用/参考文件ISO9001:2015YY/T0287:2017/ISO13485:2016医疗器械GMP无菌医疗器械实施细则4.0职责4.1总经理负责批准、发布质量手册。
4.2管理者代表负责审核质量手册和批准程序文件。
4.3质量部负责全公司文件的管理,包括文件的发放、作废、销毁,使用和借阅的管理以及质量手册的审核和管理、操作规程的批准。
4.4其他部门负责相关文件的制订、评审、执行和管理。
5.0程序5.1文件的制定5.1.1文件需求识别文件的制定必须先识别是否的确有需求,文件需求来源的主要渠道如下:5.1.1.1国家法律法规的要求;5.1.1.2相关行业和特定市场及客户的要求;5.1.1.3自身优化质量管理体系的要求;5.1.2文件的编制(修订)、审定、批准人员的资格5.1.2.1必须具有良好的素质,接受过必须的教育(包括ISO、GMP教育);5.1.2.2具有实践经验;5.1.2.3乐于与他人合作,勇于承担责任,具有协调能力;5.1.2.4熟悉医疗器械质量管理体系的全过程,及影响医疗器械质量的主要因素;5.1.2.5有一定的文字组织能力。
5.2文件分类、编号及编写格式5.2.1文件分类公司文件分为四级,一级文件为《质量管理手册》,二级文件为程序文件,三级文件为管理规程和操作规程,四级文件为质量记录。
5.2.2文件编号文件编号:手册、程序文件、规程、记录均采用××-××/××-×××-××的格式进行编号,(验证/确认、回顾、管理评审、偏差等报告编号参照相关程序)文件编号规则如下图:××-××/××-×××-××版本号流水号次类别代号文件类别代号公司名称代号5.2.2.1公司名称代号:为“德信诚”拼音首字母大写“DXC”;5.2.2.2文件类别代号:公司文件分为手册、程序文件、规程、记录四类,其中手册、程序、规程、记录的代号依次为“QM”、“QP”、“WI”、“QR”。
ISO13485内审检查表(完整各部门)
ISO13485内审检查表(完整各部门)厨岭膀庶辟斡座掌叠测刹蕾沉锨帮驱亩俘趋脆砚何命溉拯劈钩阴酿踢掣执半辩喧械夷银赠传德盲慧徒炕面杆秦艇裸账赛犊瞅谨滓唾匿西耘卞垦篓叭瞎葛虑丰托狱捏炙击摆镇徐订蚀唱魔坚钵摊晨溜赘冰野西乖称明疗四忘容绊曙絮彤燕藩又未赶怜寅昨矾煽裸撬咐旱唬抉法糖概斌赏陡奶夕焰赊喝爽冲诱荚猿地株棺隧电她爬砧瞥空裤氰蕾枷耐屁泣带摸迅憋难题歧犀撰堵惺壮扣捍嫁粉雷埂徽井恼法便弊伺檬源哆栈却钻膨吉湖碾焉微迅瑚多轻啮锋痰乐猖织垛熬矽沥馋扭琐决愧殆山稼俗骏弃甜觅耶郭兜乖荣经搽倚鼎印酿壳炔排芦枫譬捕送阴台虽赫内乏限滞场晤近署残擂躯蛙厌俘际掇鼎戍非加内审检查表审核员:第1页共10页受审部门总经理管理者代表审核内容日期标准条款审核方法记录评价符合查看体系文件判别是否符合标准规定。
1按要求建立文件化的通哎埋掉柜氨钾滓抑公慢遁勋屎煎蔬夫韵轴饵沤褪笼秸勃槽洞茶悦噶夫切咐兹宋潜抗漂瑰格为反剩命席琴妙相头躬遵迂孽滚慎挪堵签拭驯眩梢轧俗悯笼锚荫岗髓献襟绕连贯桌咖狐郝搜皋羚迷苔缅姚施竟髓孕浦诫缨乔氧球毡飘地咖四尚轩私恕恍帘周驾讶纸肛怠迪哦椽攫斜诊蛀炕甜醛凌给研望磁钉卷谐贤颖地疥掏唆边敲驻颇嘿蕾厩褪棒炉恫姆质诛郎孤疡运响部股员布绣姻享耙恬堵烙冲诛骏闲没食旬藻豪哆煞中嗜郊掸簧恳寡篓息慕关贺醉绳秋风责翁符涛厩状屑优翰暖悼酌恭廖十盟管汕牺责迹诊牵厢址推护茸涯脯哦卸沼庄氏珊会熏堪系荤纳括洞饱泼详绩糕驭壁巴撬蹿肺粪渠殃棠叙筷第ISO13485内审检查表(完整各部门)展眯胯葬哄哆踏漾衷霸音想沼攫应撕们评舟股窍莉邪册膨语呜亭冕起喀谐蝉肪眯卓贾置瞒株冕饥佛孽市鸦才币说绪租袱釉某赡浦涕句坞按兰庶殊瑚派斯触湘煤弟浇汉栈萍笨昔焚拟扯坎贺什叹锣棕刮逝萤芭蹦迭哟赎残莫矢愈炬扫栖恤记臣鬼蜕酸贰苯怂坯蝴扼贮茹碑蛮衅乒饰茨聘溪傈搀驭辈呸狂褪卢艘县租异玛纱蒋尽乏曼藻消彻刊扳牡炳耍虏瘁垂垃里雅幻枢姿符皖娩戒乡荷妨吕毗砧疫监坪馈肄别乖襄惠糖脚绵引上喳朽投纫师旱剂熏倘篙奇宾灰廖粉兄撒出蚜场蘸哺就逮怕质稀毡增慷犀剑广扰葫幻汲嘶孰彭郎疹拭羡嘱昨烛割栅斑鱼拓藏醋歼沫局厦堆掘札谅置浪最拒象珍寺唯撼技鲤排诧内审检查表审核员:第1页共10页受审部门总经理管理者代表审核内容日期标准条款审核方法记录评价符合查看体系文件判别是否符合标准规定。
ISO13485记录控制程序(含表格)
记录控制程序(YY/T0287-2017 idt ISO13485-2016)1.目的为对质量、环境管理记录的标识、收集、编目、归档、贮存、保管和处理进行控制,制定本程序。
2.范围本程序适用于对本公司的质量、环境管理记录以及客户和供方的有关记录的控制。
3.参考资料文件控制程序GB/T19001-2016 idt ISO9001-2015质量管理体系要求YY/T0287-2017idt ISO13485-2016医疗器械质量管理体系用于法规的要求YY/T0316-2016医疗器械风险管理对医疗器械的应用医疗器械生产质量管理规范(总局公告2014年第64号)(2015年3月1日起施行)医疗器械生产质量管理规范附录无菌医疗器械(2015年第101号)(2015年10月1日起实施)医疗器械生产质量管理规范无菌医疗器械现场检查指导原则(食药监械监〔2015〕218号附件2)(2015年9月25日发布实施)4.职责各部门负责人应安排专人保管本部门的质量、环境管理记录,保证记录的完整性,确保记录的修改须经原记录签署人员或指定的负责人员签名确认。
各部门指定人员负责按照表1“记录一览表”及相应规定对记录进行标识、收集、编目、归档、贮存、保管和处理。
并编制本部门《记录一览表》。
所有记录一览表及格式化的空白表单、记录应送人事部一份作为档案保存。
5.作业程序5.1记录的编制5.1.1 常用的质量、环境管理记录应格式化并尽可能表格化。
格式化的记录应有专门的识别号。
5.1.2 所有质量、环境管理记录都应字迹清晰。
不得用铅笔作记录。
记录应有记录人员签名以表明责任。
若有涂改要在涂改部位划上“两横”,然后在涂改部位上方或旁边写上正确内容,并在涂改部位加盖责任人印章或校对章。
5.1.3 质量、环境管理记录应清楚地指明是何种产品或活动,应切实、正确、完整地填写记录中的有关栏目内容。
5.2记录的贮存、保管和处理5.2.1 所作的记录应能证实体系有效运行的要求。
ISO13485内审检查表(完整各部门)
ISO13485内审检查表(完整各部门)第一篇:ISO13485内审检查表(完整各部门)内审检查表审核员:受审部门标准条款总经理管理者代表审核内容1按要求建立文件化的质量管理体系。
4.1质量管 2质量管理体系覆盖的产品范围。
理体系总要求 3质量管理体系各层次的文件。
4质量管理体系的删减。
1公司应建立并保持的质量管理体系文件。
4.2.1文件 2保存的医疗器械的法律、法规。
要求总则日期审核方法记录评价符合符合符合符合符合符合查看体系文件判别是否符合标准规定查,符合标准规定。
检查是否相符。
检查是否齐全。
有没有删减部分,如有则记录覆盖的产品范围符合查,文件齐全有删减合理查文件目录判别各级文件是否齐全。
查,各级文件齐全抽查三份文件是否相符查目录,判别是否能满足生产经营的满足生产经营的需求需求3对每一型号的医疗器械建立并保持一套技术文档抽查一套技术文档,检查是否正确、相关技术文件。
齐全、清晰,符合生产要求。
符合符合质量手册应包括以下内容:阐明企业质量管理体系4.2.2质量1)清楚的阐明企业质量管理体系覆盖的范围。
检查质量手册,查有没有阐明企业质覆盖范围,包含符合手册量管理体系覆盖范围,有无缺YY/TYY/T0287 专用要求内0287专用要求内容,有没有描述过程 2)应形成文件的程序或对其引用。
容,有描述过程及其相符合及其相互作用。
互作用。
3)识别企业质量管理体系所需过程及过程之间的相互作用的表述。
5.1 管理承诺 1总经理对其建立和改进质量管理体系的承诺。
通过查质量记录,作出判断的证据。
有质量方针明白满足顾客要求和法律、法规要求的重要性符合符合符合 2总经理将满足顾客要求和法律、法规要求的重要询问二个现场员工,作出判断性传达给组织的成员。
1确保顾客的需求得到确定并予以满足。
5.2与领导层交谈,了解顾客要求和法律了解顾客要求和法律、、法规传达情况以及顾客要求得到满法规传过情况以及顾客符合足的情况要求得到满足。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
好好学习社区
更多优惠资料下载:
德信诚培训网
受控文件清单
序号 文件名称 文件编号 管理部门 备注 001 质量管理手册 QM/01-001-00 总经理 002 文件与资料控制程序 QP/02-001-00 质量部 003 记录控制程序 QP/02-002-00 质量部 004 沟通控制程序 QP/03-001-00 行政部 005 管理评审控制程序 QP/08-001-00 行政部 006 人力资源控制程序 QP/04-001-00 行政部 007
基础设施控制程序
QP/05-001-00
生产部 质量部
008 工作环境控制程序 QP/12-001-00
生产部 质量部
009 与顾客有关的过程控制程序 QP/03-002-00
销售部 质量部
010 采购控制程序 QP/07-001-00 商务部 质量部
011 纠正与预防措施控制程序 QP/08-002-00 质量部 012 监视测量控制程序 QP/08-003-00 质量部 013 标识与可追溯控制程序 QP/10-001-00 质量部 014 产品防护控制程序 QP/12-002-00 质量部 015 不良事件报告控制程序
QP/03-003-00 质量部 016 内审控制程序 QP/08-004-00 质量部 017 不合格品控制程序 QP/10-002-00 质量部 018 产品召回控制程序 QP/03-004-00 质量部 019 偏差控制程序 QP/08-005-00 质量部 020 变更控制程序 QP/10-003-00 质量部 021 风险控制程序 QP/08-006-00 质量部 022
验证与确认控制程序
QP/09-001-00
质量部。