ISO 15189实验室认可性能验证方案
ISO-15189实验室认可性能验证方案

医学实验室ISO 15189认可性能验证实验方案为了满足目前医学实验室认可的需求保证实验室检测结果的准确性,特制定本方案。
适用于强生VITROS产品的试验项目的性能验证,包括V250/V350/V950/FS5.1//V3600/V5600上所能开展的所有定性检测项目。
本方案从准确度、精密度、参考范围、线性范围以及方法学比对5个方面对各个试验项目进行评价。
一、精密度(Precision):精密度是指在规定条件下所获得的检测结果的接近程度,表示测定结果中随机误差大小程度的指标。
精密度通常用不精密度表示。
可以分别评价连续精密度(批内精密度)、重复性不精密度(中间精密度,包括批间、日间精密度等)和再现性精密度。
本方案采用批内和天间两种方法对各个试验项目的精密度进行评价。
全部实验过程使用同批号试剂和质控品,并且保证检测当日质控在控。
1、批内精密度(连续精密度):方法:在检测患者标本过程中,连续运行高低水平质控品各20次,记录检测结果。
计算批内精密度的CV值和SD值。
结果评价(1)厂家评价标准:计算精密度指数=验证SD/厂商SD,精密度指数要求小于等于1,或者实测CV小于等于厂家要求的CV,两者符合其一即可。
具体见《精密度评价》表格。
(2)按照国际推荐标准:批内精密度应在CLIA88允许误差的1/4以内,见美国CLIA’88能力比对检验的分析质量要求。
2、天间精密度(中间精密度):方法:同样使用两个水平的质控品,若需复溶冻干质控品做实验,要注意选择产品的稳定性和瓶间差。
要严格控制每次复溶冻干品时的操作手法。
连续测试20天,每天检测1次。
在次过程中不能更换试剂批号及质控品批号,是否需要重新定标则取决于实验室。
测试完成后记录检测结果。
结果评价(1)厂家评价标准: 计算天间的SD及CV值,并计算精密度指数=验证SD/厂商SD。
精密度指数要求小于等于1,或者实测CV小于等于厂家要求的CV,两者符合其一即可。
具体见《精密度评价》表格。
最新iso15189:新版医学实验室认可规范

ISO15189:2012的新变化 2、主要修订内容概述
(3)条款框架
新版的很多条款中增加了“总则”小条款;原版中 部分小条款内容被合并为段落,新版中许多要求在被 分成小条目,清晰排列。
ISO15189:2012的新变化 3、具体变化
(1)引言
--新版的引言中,对于标准的适用范围增加了“临 床生理学、医学影像学和医学物理学”。从中可以看 出,新标准的适用范围有扩展。
理解:在新版中去掉“专用”说明标准的通用性更强,表现在新标准的
适用范围和标准的使用方更加广泛,其中适用范围明确增加了“临床生理 学、医学影像学和医学物理学”等学科,使用方除了认可机构外,增加了 “实验室客户和监管机构”。
ISO15189:2012的新变化 2、主要修订内容概述
(1)整体编排:--变动较大
➢ ISO15189:2007
全称: Medical laboratories-Particular requirements for quality and competence(医学实验室质量和能力的专用要求)
➢ ISO15189:2012
全称: Medical laboratories-Requirements for quality and competence(医学实验室 质量和能力的要求)
解读:标准的使用方除“认可机构”外,增加了“实验室客户和监管机
构”,从而使标准的应用面更广泛。增加“注”,说明医学实验室应该 在遵守相关法律法规的前提下开展相关工作。
ISO15189:2012的新变化 2、主要修订内容概述
(3)术语与定义
ISO15189:2007
3.1 认可 3.2 测量准确度 3.3 生物参考区间 3.4 检验 3.5 实验室能力 3.6 实验室负责人 3.7 实验室管理层 3.8 测量 3.9 医学实验室 3.10 检验后程序 3.11 检验前程序 3.12 原始样品 3.13 量 3.14 质量管理体系 3.15 委托实验室 3.16 样品 3.17 溯源性 3.18 测量正确度 3.19 测量不确定度
微生物检验15189认可

ISO15189实验室认可中临床微生物检验要点举例分析首都医科大学附属北京友谊医院临床检验中心苏建荣主要内容Guidance in the Field of Clinical Microbiology痰涂片:质量(回答问题),既是人员也是检验前在生物安全柜中(现场检查),也是安全Hodge试验(霍奇试验)常规?M ulti-D rug R esistanceMDR什么是MDR/PDR?摘自天津胡志东稿什么是MDR/PDR?MDR(多重耐药)指细菌对包括头孢菌素类、青霉素类、喹诺酮类、氨基糖甙类、碳青霉烯类等在内的抗生素中的至少2-3类耐药。
PDR(泛耐药)指细菌除对粘菌素、舒巴坦可能敏感外,对临床上常见的抗生素全部耐药,PDR是MDR中的特殊类型。
PDR产KPC酶(A类碳青霉烯酶)细菌的检测◆对一种或多种头孢菌素碳青霉烯类(续)对由这些分离菌引起的感染患者,应通知临床经治医师和感染控制人员,考虑选用其他抗菌药物。
初筛试验--2009、2010初筛试验--2011天津医科大学总医院⏹执行Modified Hodge Test (MHT)试验时机?⏹初筛试验阳性和对III亚类头孢菌素(如头孢哌酮、头孢噻肟、头孢他啶、头孢唑肟和头孢曲松)中一种或多种药物耐药改良Hodge试验耐药基因检测PCRTaq基因检测结果M 1 2 31500900700500400200100920bp注:M 为DNA marker, 1.KPC 阳性对照菌;2.临床分离肺炎克雷伯菌;3.阴性对照ATCC25922图2肺炎克雷伯菌bla KPC-2基因PCR 扩增产物电泳图小结:纸片初筛阳性5.2 设施环境5.2.1.1 实验室设施符合等级要求要有评估评估程序及依据生物安全(运送与交接)5.2.1.2 空间足够(领导)分子生物学要有独立设置三区5.2.2 闭门器洗眼设施有消毒及放水日期5.2 设施环境5.2.4 照明及电源(UPS)5.2.5 温湿度(记录)(校准的修正因子)5.2.10 病原存放遵守安全条例专人、专地、专记录设备5.3.1 设备配置(*关键技术要求评审原则) 无菌体液的直接显微镜检查应具备细胞离心机。
ISO15189性能验证个人经验分享 ZT

的均值及CV,以CV≤20%时的最大稀释管浓度作为可报告范围的低限;高值样 本按照一定比例稀释后的系列样本都重复测定3次,计算回收率R=测定均值/理 论值,回收率R在80%-120%之间的最大稀释倍数即为该方法的最大可稀释倍数, 可报告范围上限 = 最大可稀释倍数×线性范围上限。
7、参考文献
1、 CNAS-CL02-A003:2018《医学实验室质量和能力认可准则在临床化学检 验领域的应用说明》
注1:通常较高值样本的不精密度较小,较低值样本的不精密度偏大。对低值有临床意 义的检测项目,宜评估有判断价值的低水平样本的不精密度。 注2:如检测结果没有明确的医学决定水平,可在参考区间上限左右选一个浓度,再根 据检验项目的特点在测量区间内选择另一个浓度。 注3:如与厂商或文献报导的不精密度比较,所选样本水平宜与被比较的样本水平接近。
4、线性区间ቤተ መጻሕፍቲ ባይዱ
1.2、验证方法及结果判断 方法:每个浓度水平的样本重复测定3~4次。所有样本应在一次运行中
或几次间隔很短的运行中随机测定,最好在1天之内完成。 判断:免疫法:相关系数r2≥0.95,a在0.95-1.05范围内,截距b与0无显著
差异;酶法: r2≥0.99 , a在0.97-1.03范围内,截距b与0无显著差异。或者按照 WS/T 408-2012文件要求做线性回归分析和线性检验。
可报告范围高限验证(最大可稀释倍数):使用混合血清或5%牛血清白 蛋白生理盐水溶液或测定方法要求的稀释液对高值待测样本(必要时可添加被 分析物,并计算出理论值)进行一系列稀释,并记录稀释倍数。
6、临床可报告范围
1.2、验证方法及结果判断 在一次运行中将每个低值样本重复测定5~10次,分别计算每个低值样本
4、线性区间
最新iso15189:新版医学实验室认可规范
ISO15189:2012的新变化 2、主要修订内容概述
➢ ISO15189:2007
全称: Medical laboratories-Particular requirements for quality and competence(医学实验室质量和能力的专用要求)
➢ ISO15189:2012
全称: Medical laboratories-Requirements for quality and competence(医学实验室 质量和能力的要求)
4.14.3 内部审核的结果应提交实 验室管理层进行评审。
ISO15189:2012
修改部分: 1、标题变更为“评估和审核”,
增加“总则”。 2、增加了关于“评估”的内容,
包括“申请、程序和样品要求 适宜性的定期评审、用户反馈 的评审、员工建议、风险管理、 质量指标”等方面。 3、对于“审核”,除内审外, 还增加了外部机构的审核。除 了“内部审核”,各类评估活 动能够从多角度、多维度对实 验室管理体系进行评价,及时 识别改进机会。
ISO15189:2012的新变化 3、具体变化
(4)管理要求
11)预防措施:新版中不再分小条款,并对预防措施程序规
定了详细要求,有利于指导实验室主动及时采取有效的预 防措施,防2)持续改进:原版的5个小条款在新版中合并,并增加相
关要求。“风险管理”是新版标准引入的一个方法,将其 作为管理工具应用于持续改进,将更加有效地针对实验室 管理体系中的“短板”进行防控,达到体系自我完善的目 的。
ISO15189体系性能验证报告模版-EP15

天冬氨酸氨基转移酶(AST)检测性能验证1 目的验证AST检测性能是否满足临床要求,满足客户需求。
2 材料2.1 仪器日本TOSHIBA公司TBA-120FR型全自动生化分析仪。
2.2 试剂湖南圣湘AST(速率法)试剂,朗道多项生化校准品水平2、BIO-RAD质控品水平1、水平2。
3 方法3.1 精密度参考EP15-A《用户对精密度和准确性能的核实实验-批准指南》;允许总误差参考《卫生部临床检验中心室间质量评价标准》,采用2个浓度的样本(质控物),每天分析1批,每批对2个浓度样本重复测定4次,连续测定5天。
统计数据,计算批内精密度(Sr)和实验室内精密度(总精密度)(Sl),进一步计算批内CV(%)、总CV(%),与允许范围比较,判断验证是否通过。
页脚内容1页脚内容2页脚内容3页脚内容4页脚内容53.2 正确度采用室间质评回报结果作为评价指标。
页脚内容63.3 参考区间取40个健康人群的检测数据,其中有不超过4个数据落在参考区间以外,则可确定该项目的参考区间。
页脚内容7页脚内容8页脚内容93.4 分析测量范围取评价项目的高、低值病人标本各一份,(高值标本为接近线性范围上限或高于线页脚内容10性上限10%~15%的标本,低值标本为接近线性范围下限的标本)。
低值标本为 1 号,高值标本为6 号,低高值标本4:1 混和为2 号,3:2 混和为3 号,2:3 混和为4 号,1:4混和为5号。
每个编号标本重复测定2次,实际测定值/理论值×100%在90-110%之间均可以接受。
经统计后拟合回归线Y=bX+a。
判断标准:相关系数R2>0.95,b在0.97-1.03范围;在直线回归方程可接受基础上,如果验证线性范围小于厂家申明的线性范围,是由于没有找到更高浓度的标本,没有验证全部线性范围,将验证的上限扩展为(上限+上限*10%),验证下限扩展为(下限-下限*50%);若验证上限超出厂家声明上限,以厂家线性范围上限为准,若没有超出厂家声明上限,以自行验证为准。
医学实验室ISO15189认可性能验证实验方案

医学实验室ISO15189认可性能验证实验方案
1.引言
引言部分应该简要介绍实验室的背景和目的。
解释为什么需要进行性能验证实验,并描述该实验对实验室的重要性。
2.目标
明确性能验证实验的目标。
例如,验证分析方法的准确性、精确性、灵敏度和特异性,以及实验室是否能够在正常操作条件下提供可靠和一致的结果。
3.实验原则
描述性能验证实验的基本原则。
例如,实验应在真实且代表性的样本中进行,应根据适当的统计方法进行数据分析,结果应与已建立的参考标准进行比较,等等。
4.实验设计
详细描述实验设计。
实验设计应包括实验样本的选择和样本数量的确定。
样本应具有广泛的范围和不同的特征,以便能够提供对实验方法的全面评估。
5.实验步骤
详细描述性能验证实验的实施步骤。
每个步骤应包括所需的试剂、设备和仪器,以及步骤的详细说明。
这些步骤可能包括样本的准备、分析方法的执行、数据记录和分析等。
6.数据分析
描述实验数据的分析方法。
分析应包括对数据的描述性统计分析、准确性和精确性的计算、结果的比较和解释等。
7.结果和讨论
描述性能验证实验的结果和讨论。
结果应与已建立的参考标准进行比较,以评估实验方法的准确性和可靠性。
讨论应分析实验结果并解释其意义,讨论实验过程中可能出现的问题,并提出改进措施。
8.结论
总结性能验证实验的结果和结论。
结论应明确实验方法的准确性和可靠性,并提出改进实验方法或流程的建议。
15189认证仪器项目性能参数验证简要方案
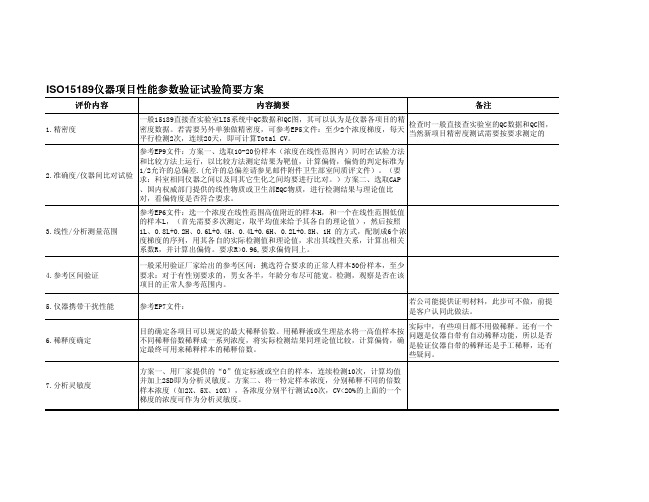
评价内容
1.精密度
内容摘要
备注
一般15189直接查实验室LIS系统中QC数据和QC图,其可以认为是仪器各项目的精 检查时一般直接查实验室的QC数据和QC图, 密度数据。若需要另外单独做精密度,可参考EP5文件:至少2个浓度梯度,每天 当然新项目精密度测试需要按要求测定的 平行检测2次,连续20天,即可计算Total CV。
参考EP9文件:方案一、选取10-20份样本(浓度在线性范围内)同时在试验方法 和比较方法上运行,以比较方法测定结果为靶值,计算偏倚,偏倚的判定标准为 1/2允许的总偏差.(允许的总偏差请参见邮件附件卫生部室间质评文件)。(要 2.准确度/仪器间比对试验 求:科室相同仪器之间以及同其它生化之间均要进行比对。)方案二、选取CAP 、国内权威部门提供的线性物质或卫生部EQC物质,进行检测结果与理论值比 对,看偏倚度是否符合要求。 参考EP6文件:选一个浓度在线性范围高值附近的样本H,和一个在线性范围低值 的样本L,(首先需要多次测定,取平均值来给予其各自的理论值),然后按照 1L、0.8L+0.2H、0.6L+0.4H、0.4L+0.6H、0.2L+0.8H、1H 的方式,配制成6个浓 度梯度的序列,用其各自的实际检测值和理论值,求出其线性关系,计算出相关 系数R,并计算出偏倚。要求R>0.96,要求偏倚同上。 一般采用验证厂家给出的参考区间:挑选符合要求的正常人样本30份样本,至少 要求:对于有性别要求的,男女各半,年龄分布尽可能宽。检测,观察是否在该 项目的正常人参考范围内。 参考EP7文件: 若公司能提供证明材料,此步可不做,前提 是客户认同此做法。
7.分析灵敏度
3.线性/分析测量范围
4.参考区间验证
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
医学实验室ISO 15189认可性能验证实验方案为了满足目前医学实验室认可的需求保证实验室检测结果的准确性,特制定本方案。
适用于强生VITROS产品的试验项目的性能验证,包括V250/V350/V950/FS5.1//V3600/V5600上所能开展的所有定性检测项目。
本方案从准确度、精密度、参考范围、线性范围以及方法学比对5个方面对各个试验项目进行评价。
一、精密度(Precision):精密度是指在规定条件下所获得的检测结果的接近程度,表示测定结果中随机误差大小程度的指标。
精密度通常用不精密度表示。
可以分别评价连续精密度(批内精密度)、重复性不精密度(中间精密度,包括批间、日间精密度等)和再现性精密度。
本方案采用批内和天间两种方法对各个试验项目的精密度进行评价。
全部实验过程使用同批号试剂和质控品,并且保证检测当日质控在控。
1、批内精密度(连续精密度):方法:在检测患者标本过程中,连续运行高低水平质控品各20次,记录检测结果。
计算批内精密度的CV值和SD值。
结果评价(1)厂家评价标准:计算精密度指数=验证SD/厂商SD,精密度指数要求小于等于1,或者实测CV小于等于厂家要求的CV,两者符合其一即可。
具体见《精密度评价》表格。
(2)按照国际推荐标准:批内精密度应在CLIA88允许误差的1/4以内,见美国CLIA’88能力比对检验的分析质量要求。
2、天间精密度(中间精密度):方法:同样使用两个水平的质控品,若需复溶冻干质控品做实验,要注意选择产品的稳定性和瓶间差。
要严格控制每次复溶冻干品时的操作手法。
连续测试20天,每天检测1次。
在次过程中不能更换试剂批号及质控品批号,是否需要重新定标则取决于实验室。
测试完成后记录检测结果。
结果评价(1)厂家评价标准: 计算天间的SD及CV值,并计算精密度指数=验证SD/厂商SD。
精密度指数要求小于等于1,或者实测CV小于等于厂家要求的CV,两者符合其一即可。
具体见《精密度评价》表格。
(2)按照国际推荐标准:批内精密度应在CLIA88允许误差的1/3以内,见美国CLIA’88能力比对检验的分析质量要求。
二、准确度准确度(accuracy)指检测结果与被测量物真值之间的接近程度。
是分析测量范围、分析灵敏度以及生物参考区间评价的基础。
准确度的评价方法很多,比如检测定值参考物质,同参考方法进行比对,同有溯源性的检测系统进行方法学比对,卫生部临检中心质评的汇报结果均可以作为评价准确性的方法之一。
本方案采用测定定值标准物质的方法来评价各个检测项目的准确度。
定值标准物质采用厂家定值标准品。
方法:(1)试验期间保证机器状态正常,保证试验当日室内质控在控。
(2)按厂家要求准备各个项目的新批号定标品(要与定标时使用的定标品批号不同)各一套,按照标准复溶。
(3)记录该新批号定标品的定值。
(4)在设备上检测各个定标品(多水平)的相关项目,每个水平重复2次,记录检测结果,将检测结果录入《准确度评价》表格。
结果评价:(1)厂家标准:按照各定标品各水平的定值和不确定度(厂家提供),来确定准确度的偏倚范围。
计算实测均值的各项目各水平定标品的偏倚,与偏倚范围相比较,来判断偏倚是否可以接受。
详见《准确度评价》表,实测的均值如果落在该限度内,则为其准确度认为可被接受。
(2)按照国际推荐标准:准确度偏倚应该在CLIA88允许误差的1/2以内,见美国CLIA’88能力比对检验的分析质量要求。
三、参考范围验证参考 NCCLSC28一A2文件,本方案仅对厂家提供的或者实验室正在使用的各个项目的参考范围进行验证。
方法:(1)选择20个能够代表实验室的健康总体的标本。
(2)保证试验系统运行正常,依照标准操作程序检测标本。
(3)运行该20份标本,记录结果,将结果填入《参考区间验证》表格。
结果评价如果20个参考个体中不超过2例的检测值在验证的参考限之外,厂商或提供参考区间的实验室报告的95%参考区间可以接受。
如果3例以上超出界限,再选择20个参考个体进行验证,若少于或等于2个观测值超过原始参考值,则可接受,若还有3个超出参考限,需要重新检查各种条件,决定是否建立自己的参考区间。
四、线性范围分析测量范围即定量检测项目的线性范围,是整个检测系统(包括仪器、试剂、校准品、质控品、操作程序、检验人员等)对应于系列分析浓度的仪器最终输出的信号间是否呈恒定比例的性能,是一个很重要的仪器性能分析指标。
本实验通过测定不同配比比例的高低值新鲜患者标本,以验证实测值和理论值的线性关系,来评估每个检测项目的线性范围。
方法:(1)实验室人员必须十分熟悉仪器的操作、质量控制和定标方法,以及正确的收集样本。
试验期间保证仪器状态良好下,质控在控。
(2)全部实验数据尽可能在较短的时间内收集,如可能,单个分析试验最好在一天内做完。
(3)用于验证线性范围的标本类型应与临床测试所用的标本类型相同或相类似,所有标本应不含厂家所标定的干扰因素(如溶血、黄疸、脂血等)。
如果上述条件不可避免,则应在最后的报告中注明在评价实验中所用的标本处理方法或基质类型。
注意:收集的高浓度标本应尽可能的接近线性范围高限。
(4)按照《线性评价》表格要求配比标本。
将H 和L 样品按: 5L 、4L+IH 、3L+2H 、2L十3H 、lL十4H 、5H 关系各自配制棍合,形成系列评价样品。
(5)难以收集到低限样品,可收集高值样品,用相应稀释液作系列不同程度稀释,形成系列评价样品。
(6)在《线性评价》表格记录检测结果。
结果评价:将数据填入《线性评价》表格,以X表示各样品的预期值,以Y表示各样品的实测值,得出散点图。
若所有实验点呈明显直线趋势,用直线回归对数据进行统计,得直线回归Y=bX+a,若r2>0.95,斜率b在1.00±0.03范围内,可直接判断测定方法在实验所涉及的浓度范围内成线性。
五、用患者标本进行方法比对及偏倚评估实验室准备用一个新的检测系统或测定方法(或新的试剂盒、新仪器进行病人标本测定前,应与原有的检测系统或者公认的参考方法-起检测一批病人标本,从测定结果间的差异了解新检测系统或方法引人后的偏倚。
如果偏倚不大,或者偏倚量在允许误差范围内,说明两检测系统或方法对病人标本测定结果基本相符,新检测系统或方法替代原有检测系统或方法不会对临床引人明显偏倚,这样的实验称为方法学比较实验。
在方法学比较中,常将新方法称为实验方法,与之比较的方法称为比较方法。
试验方法:(1)各种仪器处于良好的工作状态,严格按SOP操作。
(2)检验人员有足够的时间熟悉检测系统的各个环节,熟悉评价方案。
(3)在整个实验中,保持实验方法和比较方法都处于完整的质量控制之下,始终对实验结果有校准措施。
(4)实验时间至少做5天,时间长一些更好,可以客观反映实际情况。
(5)至少做40份病人标本,多一点更好。
(6)尽可能使50%的实验标本分析物的含量不在参考区间内,各个标本分析物含量越宽越好。
(7)不要使用对任一方法有干扰的标本。
(8)每份标本应有足够的量,以便使实验方法和比较方法都能做双份测定。
例如,第l次序号为1 、2..3..4 、5..6 、7 、8 ,第2次序号为8 、7 、6 、5 ,, 4 、3 、2...1 。
两方法都按此实验。
(9)应在2个小时内两种方法对同批标本分别开始实验,最好使用当天采集的标本。
(10)实验结束后,记录数据。
保留原始数据。
结果评价:(1)不采用已明确有人为误差的结果。
(2)将所有无明显误差的实验结果记录下来。
但是,若两种方法结果的各自差值大于任一方法的批内不精密度,应查对标本,并重新实验。
若找不出原因,应保留数据备考。
(3)整个实验一定要有内部质量控制,失控时结果必须重做。
(4)对实验数据的初步筛查:①设比较方法测定结果为X 值,实验方法测定结果为Y 值。
在《方法学比对》表格上录入检测数据,若有40个标本,则有 80个 X 和Y 的结果。
②检查每一方法内现份测定值有无离群表现,先计算每一标本每一方法成对结果的差值和差值的均值。
以4 倍的各方法差值的均值为判断限,各方法内标本的成对差值都应在限值内,说明双份测定结果符合要求。
③若原数据仅40 例病人标本的结果,剔除的数据应另做实验补上。
若有1 例以上需剔除,应检查原因是标本原因,其他数据仍可使用。
无法找出原因,则保留使用所有数据。
若最大差异超过临床允许误差,应从仪器、试剂、方法上寻找原因,停止继续实验。
(5)在《方法学比对》表格上,以X均值、Y 均值和(Y-X)、X作图,通过这两种图了解线性关系,即有无明显离群点,是否呈恒定变异等情况。
如果实验结果具良好线性关系,继续处理数据。
(6)X 、Y 关系实验点有无离群表现先看图在无明显离群点。
若无,可作以后的统计;若有,应对X 、Y 配对值作离群值计算。
将每一个标本两个方法的前后两个测定值一一对应,求出第1 个X 与第1 个Y 的差值和第2 个X 与第2 个Y 的差值,并计算出所有标本总的平均差值,以4 倍的平均差值为判断限值。
所有差值都不应超出限值。
若有,为离群点,仅一点离群点,剔除。
有一点以上离群点,需查原因,判断是否保留数据。
若原因不清,不能随意剔除,全部保留作统计分析,或者用一批新标本重做评价。
凡有剔除的,应另用标本补做。
(7)标本内分析物含量分布是否适当的检验。
相关系数r 常用来表示两个变量间互相关系密切的程度。
在直线回归统计时,除所有实验点和回归线间的离散度会影响r 值的大小外,实验点对应的分析物含量分布宽度也会明显影响r 值的大小。
若实验点过于密集,尽管离散度不大,但r 值偏小。
因此,可用r 检验X 取值范围是否适当。
一般要求r 大于或等于0.975 (或r2大于或等于0. 95) ,认为X 范围是适合的。
若r 小于0.975时,应再多做实验,扩大数据范围。
(8)线性回归统计可用直线回归分析来估计斜率和截距。
数据以回归式Y=bX十a,表示这些数据的直线趋.这是以X 方法为准,Y 方法与之配合的关系式。
式中b 为斜率,a为截距。
两方法理想状态的回归式应为Y=X,即b =1 ,a=0。
根据临床使用要求,可在各个临床医学决定水平浓度 Xc 处,了解Y 方法引人后相对于X方法的系统误差(SE) , SE == | (b -1)Xc + a |。
(9)以美国CLIA’88能力比对检验的分析质量要求允许误差的1/2为判断依据,由方法学比较评估的系统误差(SE)不大于允许误差,认为系统误差在可接受水平内。
备注:1、各个实验室应该根据自己的实际情况,建立性能验证的方法和标准。
该方法适用于科室内所有生化设备的验证。
2、上述试验方案可用于强生 Vitros 系列产品,仅供实验室参考。
参考文献:1、临床检验质量管理技术,冯仁丰,上海科学技术文献出版社2、临床检验方法学评价,杨有业张秀明,人民卫生出版社3、NCCLS, Method Comparison and Bias Estimation UsingPatient Samples; Approved Guideline—Second Edition4、NCCLS,Evaluation of the Linearity of QuantitativeMeasurement Procedures: A Statistical Approach;Approved Guideline5、NCCLS,Evaluation of Precision Performance ofQuantitative Measurement Methods; ApprovedGuideline—Second Edition。