α1受体阻滞剂
降压药物应用技巧α受体阻滞剂及中枢降压药物

降压药物应用技巧:α受体阻滞剂及中枢降压药物作者:张英来源:《中国社区医师》2010年第19期α受体阻滞剂α受体分为α1和α2受体,能同时阻断这两个受体的药物称为非选择性α受体阻滞剂,而选择性α1受体阻滞剂主要作用于α1受体,目前尚无用于临床的α2受体阻滞剂。
非选择性α受体目前有酚苄明、酚妥拉明及吲哚拉明,这类药物在降低血压的同时,可促进去甲肾上腺素释放,导致心率加快,并部分地对抗了它阻断突触后α1受体所引起的降压效应,因此,这一不足之处限制了此类药物的推广,除用于嗜铬细胞瘤引起高血压以外,一般不用于高血压患者。
选择性α1受体阻滞剂以哌唑嗪为代表,还包括特拉唑嗪、多沙唑嗪、布那唑嗪、曲马唑嗪及乌拉地尔。
这类药物对α1受体有高选择性阻断作用,不阻断突触前膜的α2受体,故减少了心动过速的发生。
其中,多沙唑嗪、曲马唑嗪较特拉唑嗪脂溶性差,与α1受体亲和力只有哌唑嗪的1/2或更少,血压下降缓和,作用时间长,直立性低血压较少,通常可维持24小时持续降压。
对于利尿剂、β受体阻滞剂、钙离子阻滞剂、血管紧张素Ⅱ受体拮抗剂(ACEI)、钙通道阻滞剂(ARB)足量应用后,仍不能满意控制血压的患者,可考虑联合应用选择性α1受体阻滞剂。
α1受体阻滞剂一般不单独用于治疗高血压。
该药的最大优点是没有明显的代谢作用,可用于糖尿病、周围血管病、哮喘病及高脂血症的高血压患者。
常见α受体阻滞剂的用量及用法见表1。
中枢降压药物传统的中枢降压药通过激动延髓α2肾上腺素受体以减少交感输出来介导抗高血压作用,20世纪60年代发现的第一代中枢降压药,如可乐定、甲基多巴等由于口干、嗜睡、阳痿、撤药后交感神经过度兴奋等严重不良反应已遭到临床医师冷落,此后发现的一些新的中枢降压药与新发现的受体,即非肾上腺能咪唑啉受体(IR)存在密切关系。
第二代中枢降压药莫索尼定和利美尼定导致的口干、嗜睡、疲惫等不良反应发生频率和严重性均明显低于第一代中枢降压药,临床报道约为10%,接近安慰剂。
αsub1sub受体阻滞剂特拉唑嗪治疗Ⅲ型前列腺炎

α1受体阻滞剂特拉唑嗪治疗Ⅲ型前列腺炎作者:宋鲁杰来源:《中国社区医师》2011年第24期前列腺炎是男性常见病、多发病,发病率高达50%。
1995年NIH提出了前列腺炎的新分类方法,将其分为4种类型,其中Ⅲ型即为慢性前列腺炎/慢性骨盆疼痛综合征(CP/CPPS)。
Ⅲ型前列腺炎是前列腺炎中最常见的类型,约占慢性前列腺炎的90%,其症状复杂多样且无特异性,病因学十分复杂,因此常常久治不愈,严重困扰男性生活质量。
目前,国内外前列腺炎诊治指南均推荐α1受体阻滞剂作为治疗Ⅲ型前列腺炎的基本药物。
近年来,特拉唑嗪作为一种长效选择性α1受体阻滞剂,如高特灵,在治疗Ⅲ型前列腺炎方面取得了良好效果,本文就此做一综述。
Ⅲ型前列腺炎临床表现Ⅲ型前列腺炎主要表现为骨盆区域疼痛,可见于会阴、阴茎、肛周部、尿道、耻骨部或腰骶部等部位。
排尿异常可表现为尿急、尿频、尿痛和夜尿增多等。
由于慢性疼痛久治不愈,患者生活质量下降,并可能有性功能障碍、焦虑、抑郁、失眠、记忆力下降等。
Ⅲ型前列腺炎治疗进展目前,越来越多的观点认为慢性前列腺炎的治疗是以缓解疼痛、改善症状、提高生活质量为目的的综合治疗。
临床治愈目前不是一个现实的目标,所以控制症状就显得尤其重要。
尹航等应用特拉唑嗪治疗ⅢB型前列腺炎患者46例,2 mg/日,连服4周。
研究结果显示,特拉唑嗪治疗后患者前列腺液中前列腺素E2(PGE2)水平(PGE2表达与前列腺炎严重程度密切相关)显著下降(P<0. 05);治疗后NIH-CPSI评分亦显著下降(P<0. 05)。
这一研究结果提示特拉唑嗪通过减少尿液反流,阻断了前列腺液中炎性因子介导的炎性反应,从而发挥治疗的功效。
在一项近期的研究中,美国华盛顿大学医学中心的Lee等12荟萃分析了10项α1受体阻滞剂治疗CP/CPPS的临床研究资料。
分析了多种选择性不同的α1受体阻滞剂的治疗效果,包括特拉唑嗪(α1受体阻滞剂),多沙唑嗪(α1受体阻滞剂),阿夫唑嗪(α1受体阻滞剂),坦索洛新(α1A受体阻滞剂)和萘哌地尔(α1D受体阻滞剂)。
α1肾上腺素能受体阻滞剂与男性性功能关系的研究进展

和研制起效快、疗效确切和毒副作用小的药物,具
有十分重要的性功
能障碍 男性
中图法分类号R 698 Q 425
参考 文 献
In:Lipshultz L1,Howards SS.Infertility in male 2nd ed.
St.Louis:Mosby.Year Book.199l:155.176
万方数据
嗪组ED发生率较低(2.8%),而安慰剂组为5%。 Kaplan等睇刮在灵长目动物猴子实验中发现多沙唑嗪 能作用于阴茎QtAR,实验中给阴茎海绵体注射多沙 唑嗪能使阴茎完全勃起。多沙唑嗪是一个高选择性Q 。AR阻滞剂,其主要作用于Q…QmAR,而在阴茎 海绵体和血管平滑肌富有此两种受体。目前推广应用的 是可多华,血药浓度稳定,副作用小。在应用可多华 治疗良性前列腺增生的临床研究中发现¨“:该药除了 能够改善良性前列腺增生引起的LUTS外,还能改 善、提高患者的性功能。Rose等¨刘在给ED病例服用 西地那非无效的情况下加用可多华作为联合用药,取 得了相当满意的效果,而且没有发生因5.型磷酸二酯 酶抑制剂与高选择性Q,AR阻滞剂联合应用而产生的 副反应。
体功能而出现的“自我保护’现象,以能经常保持阴茎海
绵体内有足够的氧饱和度。另外,去甲肾上腺素诱导的 平滑肌收缩相对于NO介导的平滑肌松弛更显著地引起 ED…1。由此,我们可以看到正确调节Q,AR功能状 态,能够改善、提高勃起功能,达到完美的性生活。
20世纪80年代初,Virag(1981)””和Brindley (1983)”w两位学者相继发现阴茎海绵体内注射罂素碱或 酚妥拉明数分钟后可引起阴茎勃起。国内姚德鸿…1在80 年代中期首先用酚妥拉明+罂粟碱联合应用于阴茎海绵 体内注射,达到了相当满意的阴茎勃起效果。酚妥拉 明是一种竞争性、非选择性a,AR和a zAR受体阻滞 药,其作用持续时间较短。通过阻断胞突接合后血管中 Q-AR和Q 2AR受体,因而引起血管内平滑肌扩张; 在阴茎海绵体局部应用使得海绵体内平滑肌松弛和动 脉血管平滑肌舒张,血流量增加。罂粟碱直接作用于阴茎 海绵体平滑肌细胞,抑制磷酸二酯酶,增加细胞内环磷酸 腺苷(cAMP)的浓度,cAMP将血管平滑肌中的触酶 钙移出细胞胞质,产生一个没有神经参与的直接的平滑 肌松弛效应,两者联合应用达到了理想的勃起效应。 1988匀ZGwinup…1首先应用酚妥拉明片剂5∞嚷治疗勃起功 能障碍,虽然能够使阴茎勃起,但是有效率较低,而 且对Q AR亚型选择性差,既有Q,AR的作用,又有Q :AR的作用。a2AR阻滞剂的口服应用对全身有一定副 作用,如:心血管系统:经常出现直立性低血压和 心动过速,偶然出现急性或延长性低血压、极少出 现心绞痛;中枢神经系统:偶然出现头晕和衰弱; 胃肠道:偶然出现恶心、呕吐和腹泻;其它方面: 偶然出现鼻塞和红晕。因此,没有得到广泛推广应 用。Grimm等“‘”’在一项5种抗高血压药物对ED的长 期影响的研究中发现:抗高血压治疗2年后多沙唑
a-受体阻滞剂

时可使去甲肾腺素释放减少,对其产生负反馈调节作用,间接影响 效应器官的反应,调节神经和组织的反应。
α-受体阻滞剂的定义
α受体阻断剂能选择性地与α受体结合,竞争性阻 断神经递质或拮抗激动剂对受体的激动作用,对β受体 基本无作用。
α-受体阻滞剂的药理作用
血管 血管扩张→外围阻力↓→血压↓
心脏 血压↓→反射性兴奋心脏:心收缩力↑ 心率↑ 心输出量↑ 阻断突触前膜α2受体,负反馈减弱→ 促进神经末梢释放NA
血压变化情况(做 酚妥拉明试验时, 在给药前、静脉给 药后至3分钟内每 30s、以后7分钟内 每1min测1次血压, 或在肌肉注射后 30~45min内每5min 测一次血压)
按照医嘱服药,如出现头晕、晕厥或 直立性低血压应如果停药几天,应重 新使用首次给药方案治疗,首次用药 在睡前,其他用药时间宜在早晨。告 知患者用该药治疗时可能出现睡意或 困倦,避免驾车,可能导致阴茎异常 勃起。
酚妥拉明 注射液
适应症
良性前列腺 增生,高血 压
诊断嗜铬细 胞瘤,治疗 左心衰及NA 外溢
常见不良反应
监护点
用药教育
无力、体位性低 血压、头晕、瞌 睡、鼻充血/鼻炎 、阳痿等
直立性低血压, 心律失常,鼻塞 ,胃肠道反应
血压变化情况,用 药后是否发生不良 反应如发生不良反 应应及时对症治疗 ,并考虑是否需要 调整剂量
忌与铁剂配伍,如出现不良反应应及 时告知医生,对症治疗。
相关药物警示信息 欧盟修改α-受体阻滞剂的产品信息 (2009年)
几种常见的α-受体阻滞剂
药名 适应症 常见不良反应
监护点
用药教育
盐酸乌拉 地尔氯化 钠注射液
高血压危象 重度、难治 性高血压用 于控制围手 术期高血压
《a受体阻滞剂》课件

儿童
儿童使用a受体阻滞剂时应根据年 龄和体重调整剂量,并密切观察不 良反应。
老年人
老年人的生理功能减退,对药物的 代谢和排泄能力下降,使用a受体阻 滞剂时应谨慎,并适当调整剂量。
05
a受体阻滞剂的发展前景
新药研发
针对新适应症的研发
03
a受体阻滞剂的不良反应
直立性低血压
总结词
直立性低血压是服用a受体阻滞剂后常 见的不良反应之一。
详细描述
当患者体位由卧位或坐位突然站立时 ,血压迅速下降,出现头晕、眼前发 黑、心悸等症状。这是因为a受体阻滞 剂会阻断血管平滑肌上的a受体,导致 血管舒张,血压下降。
心动过缓
总结词
心动过缓是a受体阻滞剂的另一个常见不良反应。
作用于血管平滑肌的a受 体,扩张血管,降低外周 阻力,从而降低血压。
抑制心肌收缩力
通过拮抗心肌细胞的a受 体,抑制心肌收缩力,降 低心输出量,从而降低血 压。
02
a受体阻滞剂的临床应用
心血管疾病
高血压
a受体阻滞剂通过抑制血管平滑肌上 的a受体,扩张血管,降低血压。适 用于轻、中度高血压的治疗。
冠心病
个体化治疗
根据患者的具体情况,选择合适的a受体阻 滞剂及剂量,实现个体化治疗,提高治疗效 果。
药物经济学评价
要点一
成本效益分析
对a受体阻滞剂进行全面的成本效益分析,评估其在不同治 疗领域的应用价值,为医疗决策提供依据。
要点二
药物经济学研究
开展a受体阻滞剂的药物经济学研究,探索其在不同国家、 地区的经济可行性,促进药物的合理使用和普及。
高血压用药

用药选择能有效控制血压并适宜长期治疗的药物就是合理的选择,包括不引起明显副作用,不影响生活质量等。
上述六类主要降压药物中:(1)合并有心力衰竭者,宜选择ACE抑制剂、利尿剂。
(2)老年人收缩期高血压者,宜选择利尿剂、长效二氢吡啶类钙通道阻滞剂。
(3)合并糖尿病、蛋白尿或轻、中度肾功能不全者(非肾血管性),可选用ACE抑制剂。
(4)心肌梗死后的患者,可选择无内在拟交感作用的β受体阻滞剂或ACE抑制剂(尤其伴收缩功能不全者)。
对稳定型心绞痛患者也可选用计通道阻滞剂量。
(5)对伴有脂质代谢异常的患者可选用α1受体阻滞剂,不宜用β受体阻滞剂利尿剂量(6)伴妊娠者,不宜用ACE抑制剂、血管紧张素II受体阻滞剂,可选用甲基多巴西(7)对合支气管哮喘、抑郁症、糖尿病着不宜用β受体阻滞剂;通风患者不宜用利尿剂;合并心脏起搏传导障碍者不宜用β受体阻滞剂及非二氢吡啶类钙通道阻滞剂。
在降压治疗过程中应遵循4项原则:(1)开始治疗时,使用小剂量药物以减少不良反应。
(2)合理的药物联合,以达到最大的降压效果。
(3)尽可能使用长效降压药,以提高治疗依从性和减轻血压波动。
(4)要求白昼及夜间稳定降压,可用动态血压方法监测。
降压目标及应用分子由于血压水平与心、脑、肾并发症发生率呈线性关系,因此,有效的治疗必须使血压降至正常范围,即降落道路140/90mmHg以下,老年人也以此为标准。
对于中青年患者(<60岁),高血压合并糖尿病或肾脏病变的患者,治疗应使血压降至130/85mmHg以下。
原发性高血压诊断一旦确立,头长需要终身治疗(包括非药物治疗)。
经过降压药物治疗后,血压得到满意控制,可以逐渐减少降压药的剂量,但一般仍需长期用药,中止治疗后高血压仍将复发。
此外,长期服药治疗者突然停药可发生停药综合征,即出现血压迅速升高,交感神经活性增高的表现如心悸、烦躁、多汗、心动过速等;合并冠心病者,可出现心肌缺血发作及严重心律失常。
(1)对于轻、中高血压患者宜从小剂量或一般剂量开始,2-3周后如血压未能满意控制可增加剂量或换用其他类药,必要时可用2种或2种以上药物联合治疗。
药理肾上腺素受体阻断药

第九章肾上腺素受体阻滞剂一、α-受体阻断药-酚妥拉明?第一节α肾上腺素受体阻断药1.按对受体的选择性:对α1和α2无选择性:酚妥拉明选择性阻断α1受体:哌唑嗪选择性阻断α2受体:育亨宾2.按作用持续时间分:短效类长效类一、短效类酚妥拉明(phentolamine)妥拉唑啉(tolazoline)药理作用1.扩张血管:小动脉、小静脉扩张→外周阻力下降扩静脉>动脉(引起直立性低血压)2.扩血管机制:阻断α1 、α2受体(为什么不作降压药使用?);直接扩张血管为什么哌唑嗪用于治疗高血压而酚妥拉明则不?3.酚妥拉明兴奋心脏:心率↑、收缩↑机制扩血管-BP ↓→反射性(+)交感神经,兴奋心脏β1受体;阻断突触前膜α2受体→NA 释放→兴奋心脏β1受体4.酚妥拉明其它作用①拟胆碱作用:兴奋胃肠平滑肌②组胺样作用:胃酸分泌、皮肤潮红③唾液腺和汗腺分泌增加酚妥拉明临床应用1.外周血管痉挛性疾病2.去甲肾上腺外漏3. 嗜铬细胞瘤:诊断、高血压危象治疗4.抗休克:扩血管作用,改善微循环5.可乐定突然停药所致高血压的抢救酚妥拉明不良反应1.拟胆碱作用:腹痛,腹泻,呕吐,诱发溃疡2.扩血管作用:低血压3.反射性兴奋心脏作用:IV时,心率加快,诱发心律失常或心绞痛。
注意事项1、缓慢注射或滴注2、胃炎、胃、十二指肠溃疡、冠心病慎用二、α-受体阻断药-酚苄明?二、长效类酚苄明(phenoxybenzamine )又名苯苄胺(dibenzyline) 体内过程及其特点1. 起效慢,氯乙胺基→乙撑亚胺基→与α-R牢固结合.2. 口服吸收少,仅作IV.(刺激性)3. 作用久:脂溶性大,缓慢释放,排泄慢,一次用药,可维持3-4天三、选择性α1 -受体阻断药–哌唑嗪?选择性α1受体阻滞药—哌唑嗪(Prazosin)用途:1.治疗高血压2.治疗前列腺增生引起的排尿困难。
特拉唑嗪(Trazosin)特点:生物利用度高、作用时间长、口服吸收好。
受体阻滞剂
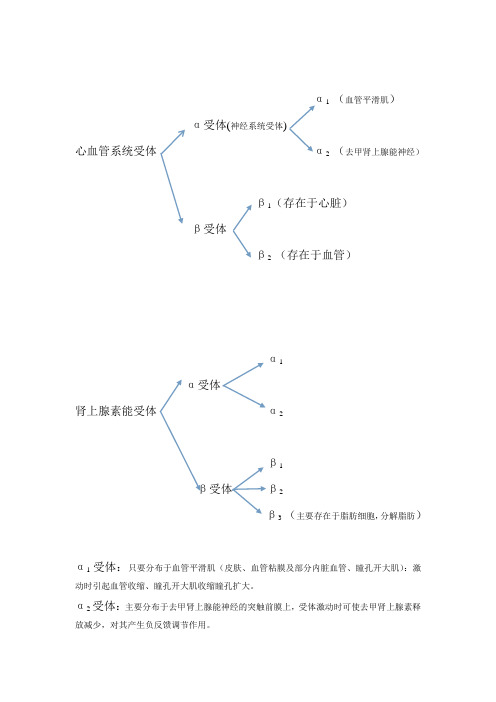
α 1 (血管平滑肌)α受体(神经系统受体)心血管系统受体α 2 (去甲肾上腺能神经)β1(存在于心脏)β受体β 2 (存在于血管)α1α受体肾上腺素能受体α 2β1β受体β 2β 3 (主要存在于脂肪细胞,分解脂肪)α1受体:只要分布于血管平滑肌(皮肤、血管粘膜及部分内脏血管、瞳孔开大肌):激动时引起血管收缩、瞳孔开大肌收缩瞳孔扩大。
α2受体:主要分布于去甲肾上腺能神经的突触前膜上,受体激动时可使去甲肾上腺素释放减少,对其产生负反馈调节作用。
血管:通过阻断血管平滑肌α1受体和直接舒张血管平滑肌作用,使血管扩张,外周阻力降低,血压下降。
心脏:由于直接扩张血管及阻断α1受体,血压下降反射性引起心脏兴奋,使心肌收缩力增强、心率加快、心排出量增加。
也可以通过阻断去甲肾上腺素能神经末梢突触膜α2受体,促使神经末梢释放去甲肾上腺素引起兴奋。
β1受体:广泛存在于心脏,可激动引起心率和心肌收缩力增加。
β2受体:存在于支气管和血管平滑肌,可激动引起支气管扩张、血管舒张、内脏平滑肌松弛等。
β3受体:主要存在于脂肪细胞上,可激动引起脂肪分解。
β—受体阻滞剂作用:主要是与儿茶酚胺对β—受体起竞争性结合,从而阻断儿茶酚胺的激动和兴奋作用。
心血管系统:阻滞心脏β受体而表现为负性变时、负性变力、负性传导作用而使心率1减慢,心肌收缩力减弱,心排血量下降,血压略降而导致心肌氧耗量降低,延缓窦房结和房室结传导,抑制心肌细胞的自律性,使有效不应期相对延长而消除因自律性增高和折返激动所致的室上性和室性快速心律失常,由于可以延长房室结传导时间而可以表现为心电图的P-R间期延长。
支气管系统:β受体阻滞剂可使支气管平滑肌收缩增加呼吸动阻力,故在支气管哮喘2或慢性阻塞性肺疾病患者,有时可加重或诱发哮喘的急性发作,但是这种作用对正常人影响较少,选择性β1受体阻滞剂此作用较弱。
然而β2受体阻滞引起血管平滑肌收缩可阻止和治疗偏头痛的发作。
代谢:β受体阻滞剂可抑制交感神经所引起的脂肪分解;β2受体阻滞剂则可拮抗肝糖原1的分解。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
α1受体阻滞剂α1受体阻滞剂α-肾上腺素能受体有:突触前α2受体、突触后或血管的α1受体和两种突触后α2后体。
目前尚无可用于α临床的α2受体拮抗剂。
α1后体拮抗剂则主要用于降压治疗。
8.1 分类及用量非选择性α1受体阻滞剂酚妥拉明和酚苄明,同时具有α1和α2的阻段作用,除用于嗜铬细胞瘤引起的高血压外,一般不用于高血压患者。
各种α1后体阻滞剂的降压作用按药代动力学尚有所不同,主要有哌唑嗪、特拉唑嗪及近年来问世的多沙唑嗪、曲马唑嗪等,后者较哌唑嗪脂溶性差,与α1受体亲和力只有1/2或更少,血压下降缓和,作用时间长,直立性低血压较少。
通常为维持24H持续降压,哌唑嗪需要每12H服用一次,特拉唑嗪或多沙唑嗪只需要每日服用一次。
(1)哌唑嗪:一般认为其降压效应与氢氯噻嗪、普萘洛尔或硝苯地平等相同。
口服易吸收,生物利用度44%--70%,1-3h后血药浓度达峰值。
对充血性心力衰竭、肾衰患者药物半衰期延长。
可单用于轻中度高血压或肾性高血压。
对妊娠、肾功能不良、合并糖尿病、呼吸道疾病及前列腺肥大的高血压患者尤为适宜。
通常服用0.5-1MG/次,2-3次/D(首剂0.5MG,睡前服)连服两周,逐渐增加剂量至2-20MG/D,分服。
对重度高血压可与利尿剂、β受体阻断剂合用,但要注意调整剂量。
不良反应有直立性低血压、眩晕、昏厥、心悸及少见的头痛、嗜睡、鼻塞、乏力等,但这些常在连续用药过程中自行减少。
(2)特拉唑嗪:化学结构与哌唑嗪相似,对血管平滑肌突触后A1受体有选择性阻滞作用,但作用强度比哌唑嗪稍弱,其特点是消除T1/2较长,约12h,因此可一日给药一次。
口服吸收完全,生物利用度约为90%。
因此利于控制用药剂量,给药后1—2血药浓度达峰值,经肝代谢,胆汁排泄。
用于治疗轻中度高血压时可单用或与b受体阻滞剂、利尿剂合用。
口服1 mg/ 次,一次/d,随血压增加剂量,可用2—20mg/d。
不良反应较少,主要为眩晕、头疼、乏力、鼻黏膜充血等。
(3)多沙唑嗪:对血管平滑肌突触后A1受阻滞剂作用强度委哌唑嗪的1/2。
但作用时间较长。
通过扩张血管、降低外周阻力而使高血压患者的站立及卧位血压下降,不影响心率及心输出量,能增加肾血流,改善脂代谢。
口服易吸收,生物利用度62%—69%。
服药后3.6h血液浓度达峰值。
用于治疗轻中度高血压,可是舒张压级收缩压下降10mmHg左右,尤其适用于合并高血脂症、糖尿病、呼吸道疾病及外周血管病的患者。
口服1—16mg/次,一次/d,维持量2-4mg/d。
在一项228例高血压患者参与的阿替洛尔双盲、随机、对照试验中,服药24月未见耐劳想象,来自HALT的大型临床实验证实服用每日一次多沙唑嗪可控制血压,服用16周能明显的降低患者的白天与夜间血压,降压最大效应在上午,提示器降压效应与a-肾上腺素能张力有关。
(4)曲马唑嗪:选择性阻滞a1受体,并直接扩张血管。
还可降低肾血管阻力,对心率无影响。
高血压患者口服曲马唑嗪后,降低立位性高血压较卧位更显著、口服50mg/次,2次/d,根据血压调整剂量,可至200—350mg/d。
本药出现体位性低血压少见。
8.2作用机制A1受体阻滞剂选择性阻滞血管平滑肌突触后膜的A1受体,舒张小动脉及静脉,而心数出量略升或不变而达到降压目的。
并且本类药在降低外周血管阻力方面比B受体阻滞剂更接近生理反应。
有证据表明,节后受体对肾上腺素能神经兴奋的敏感性改变使高血压状态下交感肾上腺活性增加的总的原因。
a1受体阻滞剂抑制节后受体对儿茶酚胺的反应,可通过干预高血压发病的基本机制而降低血压,这种药物对于在小动脉结构性改变前,血管平滑肌痉挛,血管阻力增加的患者尤其有用。
8.3特点8.3.1 对于冠心病、心肌梗死有关本药对于患者心血管疾病危险性影响的资料目前尚少。
A1受体阻滞剂虽不能直接缓解心绞痛,但可通过逆转左室肥厚与降压间接改善心肌供氧。
2000年美国和加拿大报道的the antihypertensive and lipid-lowering tr eatment to prevent heat attack trial(allhat)试验的一部分比较了a受体阻滞剂多沙唑嗪与利尿剂氯噻酮对心血管疾病的作用。
实验有24335名高血压和至少一个冠心病危险因子的患者参加并随即分为多沙唑嗪组(2-8mg/d )与氯噻酮组(12.5-25mg/d)。
计划治疗4-8年。
中期随访时两组死亡率无明显差别(4-年率:9.62%和9.08%,P=0.56),多沙唑嗪组有更高的中风率和心血管疾病发病率(4-年率:25.45%比21.76,P=0.001)。
8.3.2对于血脂代谢A1受体阻滞剂无使脂类代谢恶化的副作用。
这类药物对于血脂异常的病人有其优点,长期应用可改善脂代谢,降低TC,TG,LDL-C,升高HDL-C.美国1999年报道了高血压对于血脂和脂蛋白的长期影响的研究结果,这个实验有1292名患者参加,随机使用安慰剂和以下6种降压药物之一:氢氯噻嗪、阿替洛尔、卡托普里、可乐定、硫氮焯酮和哌唑嗪。
治疗8周后,哌唑嗪的总胆固醇降低9.3mg/dl,载脂蛋白降低5.4mg/dl,与安慰剂组无明显差异。
治疗一年后,无明显血脂和脂蛋白不利的改变。
研究表明其对于血脂和脂蛋白无长期的不利改变,可安全使用。
1991年喝1993年treatment of mild hypertension (tomh)研究认为:轻度高血压患者连续使用多沙唑嗪2mg/d,4年以后,血胆固醇水平有所下降。
8.3.3对于糖代谢、糖尿病、胰岛素抵抗A1受体阻滞剂对糖代谢无不良影响,并可提高胰岛素的敏感性。
8.3.4对于代谢及电解质A1受体组织及对代谢如血尿酸等无不良影响。
但单独长期服用已导致水钠潴留。
8.3.5对于生活质量1991年和1993年Treatment OF Mild Hypertension (TOMH)研究认为,对轻度高血压患者连续使用多沙唑嗪2md/d4年以上,尽管生活质量的改善不如醋丁洛尔组,但与安慰剂组相同。
8.3.6对前列腺增生本类药物还能减轻前列腺增生病人的排尿困难症状。
例如999年美国的一项涉及2084名患者的评价降压药物a1受体阻滞剂特拉唑嗪治疗有症状的良性前列腺肥大患者的心血管安全性的回顾性研究表明:加用特拉唑嗪对于为降压治疗的病人可降低平均收缩压5.3mmhg,对于已治疗的病人降低6.7mmhg,对于进入时有高血压的未行将压治疗的患者平均将降低的收缩压分别是2.1mmhg和1.1mmhg。
加用特拉唑嗪后对于已应用利尿剂降压的患者影响最大,平均降低收缩压12.3mmhg 。
对于舒张压的影响类似。
应用特拉唑嗪对于是否以降压治疗病人的血压相关性不利事件分别为13.5%和14.3%。
因此,对于各种血压水平和降压治疗的良性前列向肥大患者加用特拉唑嗪是安全有效的。
8.4疗效受体阻滞剂有较强的减压效用,受体阻滞剂能安全有效地降低血压,适用与中、重度高血压患者。
代表性药物哌唑嗪的降压效应与氢氯噻嗪、普萘洛尔和硝苯地平等相同。
新制剂如特拉唑嗪、乌拉地尔等的作用强度比哌唑嗪稍弱,但仍有较强的降压效应,而且负作用明显减轻。
多沙唑嗪作用强度为哌唑嗪的/2。
1991年和1993年treatment of mild hypertension(tomh) 研究认为:对轻度高血压患者连续使用多沙唑嗪2mg/d4年以上,降压效果与使用其他组药物相同。
1993年与199 4年的单剂降压试验的va试验(六种降压药物与安慰剂比较试验)中,严重的男性高血压患者连续使用哌唑嗪4-20mg/d, 一年以上的治疗有效率达54%,而安慰剂组为31% ,种族和年龄对疗效无重要影响。
8.5时应证a1受体阻滞剂使用与高血压合并血脂代谢紊乱、良性前列腺肥大,维持代谢状态和糖耐量减低或糖尿病的患者。
合并脂代谢障碍或前列腺疾病的老年高血压病人可优先选用此类药物。
a-肾上腺素能阻滞剂代表性药物是哌唑嗪,有较强的降压效应。
适用与中、重度高血压患者。
作为a1和a2阻滞剂的酚苄明和酚托拉明仅用于嗜铬细胞瘤。
8.6副作用这类药物临床不够广泛,其原因主要有两个副作用;首先是体位性低血压,这在老年人更易发生。
所以必须评定站立时的血压。
万一发生血压过低,可加用多巴胺予以纠正。
老的A1受体阻滞剂如哌唑嗪首次应用可出现严重的低血压、眩晕、昏厥、心悸等(即“守剂效应”)因为此负作用(其实是直立性低血压的发生率不足1%),故要求首次剂量减半并在睡前服用。
新制剂如特拉唑嗪、乌拉地尔这一负作用已明显减轻。
另一副作用是单独服用易致水钠潴留而降低疗效,因此在临床上较少单独使用。
有些患者可有嗜睡和偶发心动过速。
长期使用还可产生耐药作用。
8.7禁忌证严重主动脉瓣狭窄、体位性低血压为本类药物的禁忌证。
由于体位性低血压对于老年人更是特殊问题,因此A受体阻滞剂也不适合用于治疗老年高血压病。
8.8联合用药A 受体阻滞剂与B阻滞剂连用效果好。
如果与钙离子拮抗剂连用可通过两种机制产生强烈的血管扩张,易导致血压过低。
但谨慎应用则有很好的协同作用。
与ACEL联用的研究较少。
酸特拉唑嗪片通用名称:盐酸特拉唑嗪片英文名称:Terazosin Hydrochloride Tablets汉语拼音:Yansuan Telazuoqin Pian【成份】本品主要成份是盐酸特拉唑嗪。
其化学名称为:1-(4-氨基-6,7-二甲氧基-2-喹唑啉基)-4-[(四氢呋喃基)碳酰基]哌嗪盐酸盐二水合物。
化学结构式(如右图):分子式:C19H25N5O4·HCl·2H2O分子量:459.93【性状】本品为类白色片。
【适应症】轻度或中度高血压的治疗,可以单独用药或与其它抗高血压药物如噻嗪类利尿剂或β-受体阻滞剂合用。
良性前列腺增生(BPH)引起的尿潴留的症状治疗。
【规格】2mg【用法用量】高血压初始剂量为睡前服用1mg(1/2片),且不应超过,以尽量减少首剂低血压事件的发生。
一周后,每日单剂量可加倍以达预期效应。
常用维持剂量为每日一次2~10 mg(1~5片)。
剂量超过20mg(10片)未见效能增强,未对40mg(20片)以上剂量进行研究。
良性前列腺增生(BPH)根据患者的反应来调整给药剂量。
初始剂量为睡前服用1mg(1/2片),且不应超过,以尽量减少首剂低血压事件的发生。
一周或两周后每日剂量可加倍以达预期效应。
常用维持剂量为每日一次5~10mg(2.5~5片)。
给药两周后症状明显改善。
到目前为止,还没有足够的数据表明剂量超过每日一次10mg(5片)会引起进一步的症状缓解。
应当采用初始剂量开始治疗并在四周后进行疗效总结。
每次调整剂量都可能发生暂时的不良反应。
如果不良反应持续存在,应考虑减少给药剂量。